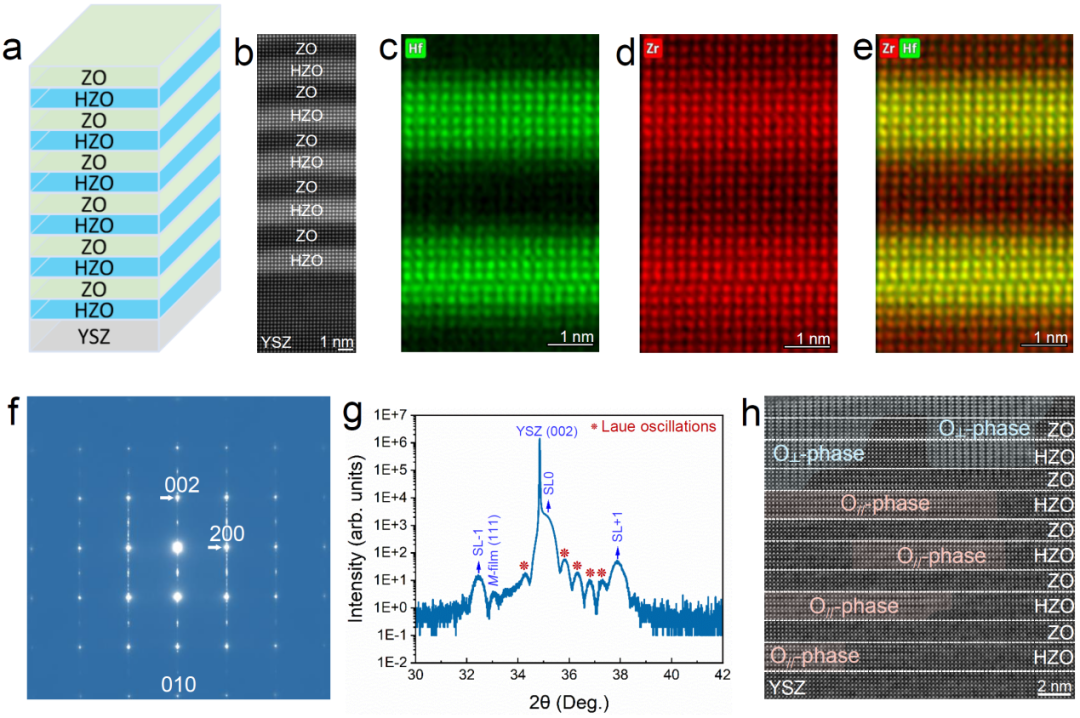
21,000㎡智能制造基地、全球超10,000个项目落地、累计出货面积突破100万㎡、单款产品销售额超5亿元——这是过去20年,深德彩在租赁与固装场景中,交出的基于数据的履历。今年,站在20周年的节点,深德彩一反往日低调常态,在短时间内连续打出三张“实牌”: 划定主线:“舞台租赁+COB专业显示+商业显示”为三大核心。 品牌分层:推出全新子品牌 SkyFlex,明确将其定位为专注高端固装领域的独立作战单元。 新品落地:针对户外及商显场景,同步迭代两款固装标杆级产品。而深德彩这次罕见地连续释放这三个明确的战略信号,这一密集动作引发了产业的高度关注。行家说Display近期调研了深德彩内部团队及多位产业人士,试图剥离营销话术,还原其2026年动作背后的考量和发展策略。20年沉淀,转为新20年的子弹根据公开资料与产业调研,深德彩在过去20年间,主要将资源集中于租赁与固装两大核心场景。除了前文提及的“21,000㎡基地、10,000+项目、近100万㎡出货量”等数据履历外,其业务足迹还涵盖了2024巴黎奥运会、APEC 2025峰会等具有全球关注度的活动。这些数据和案例的背后,并非单纯的市场扩张,而是技术与产品体系的长期迭代。■ 技术维度的“硬支撑”调研显示,深德彩累计开发了260余项专利技术,并多次获得德国Red Dot、日本Good Design等国际设计奖项。其技术护城河主要体现在两个具体维度:一是结构效率革新:例如首创的旋转式弧度锁结构,将租赁场景下的拆装操作简化为“单手2步”,并实现了±45°的高精度弧形拼接。这一设计直接量化为舞台搭建效率的提升与人工成本的降低。二是能效成本优化:DCD第三代节能技术通过动态电流驱动,使功耗与灯板温度双降20%以上,LED理论寿命延长至1.3倍。以100㎡显示屏为例,该技术在全生命周期(10年)内可为客户节省电费约28万元。这种TCO(总拥有成本)优势,成为其在商业项目中被反复选用的关键因素之一。■ 产品维度的“全球化验证”在产品层面,深德彩形成了较为清晰的双线布局:* 租赁线:提供覆盖不同创意需求的旗舰产品及配套方案,满足高频次、多变性的舞台需求。以AF V3、UL、DV2等系列为代表,分别在高刷新无扫描线成像、极致轻量化与快速维护、超高刷新率与多形态创意拼接方面形成特点。其中,DV2等系列定位“全能租赁”,以“一屏多能”的实用性著称,可灵活作为主屏、地砖屏或天幕使用。其前身D系列自2017年发布以来,已成为经久不衰的畅销型号,据称全球累计销售额已突破5亿元,并曾服务于APEC峰会、FINA世界游泳锦标赛等国际级项目,验证了其可靠性与顶级项目的适配能力。AFV3系列(马赫小子)专为高端租赁市场打造,主打精准的弧形拼接与优异的操作体验,旨在满足对舞美造型有高精度要求、对复杂创意高需求的高端租赁场景。该系列产品已成功应用于波兰电视台、秘鲁电视台项目,并参与德国、俄罗斯等多地影视拍摄制作。* 固装线:提供基于倒装COB技术和MIP技术的专业显示方案,以及全面搭载DCD节能技术的户内户外商业显示方案。满足客户在专业、商业显示产品的需求。CM系列以“达芬奇”为名,让每一处商业空间、文化公共空间化身艺术画布,自由挥洒灵感与想象。 无论标准显示还是创意显示,皆能随心呈现视频、影像与色彩,让每一处空间皆成为独特的视觉作品。真实还原万千色彩,所想即所现,让“画你想画”成为触手可及的艺术语言。ME系列(钻石系列)主打户外高亮,是深德彩近二十年户外显示经验与相关技术发展的成果,从早期的K系列演进至今,其高可靠性在深圳福田裸眼3D屏、 印度尼西亚地标性建筑广告屏等全球多地标性项目中得到印证。据悉,其固装产品亦曾服务于沙特赛马世界杯等世界级赛事,在直播严苛环境下表现获得认可,证明了其在高端固装场景下的实力。值得一提的是,深德彩的全球实践与专业资质为其产品提供了有力背书。在资质上,其已取得德国TÜV、欧盟CE、北美ETL等全球主流市场的权威认证,产品行销160多个国家与地区。在项目上,其产品不仅服务于APEC峰会、FINA世锦赛等国际盛事,更亮相于2024年巴黎奥运会、2025年APEC峰会等国际顶级舞台。这一系列从认证到高端应用的完整链条,系统性验证了其应对全球各类严苛环境与高标准需求的强大实力。正是基于上述20年的技术储备与市场验证,深德彩在2026年做出了三个看似突然、实则逻辑严密的动作。这一系列动作折射出LED显示产业竞争格局的深层变化:通用型市场的红利逐渐消退,而需求明确、技术壁垒较高的专业细分场景正在加速成熟。这些场景对供应商提出了差异化要求——不仅需要定制化的产品逻辑,更需要独立的服务体系与品牌认知。深德彩构建的“母品牌(综合平台能力)+ 子品牌(垂直领域穿透)”矩阵,或许正是应对这一市场分化趋势的理性选择。SkyFlex的设立,标志着其试图在高端固装这一“高门槛”领域,建立更精准的识别度。新20年,深德彩尝试从“做大”走向“做深”随着母公司三大主线的划定,以及子品牌SkyFlex 继2026年ISE展会全球首秀后,又在ISLE展会完成国内正式发布,深德彩的战略蓝图已从“规划期”全面转入“实战期”。当全球市场规模增速放缓,竞争焦点正从单纯的“市场份额”转向“利润质量”与“客户全生命周期价值(LCV)”。深思之下,深德彩的这一举措,或也为同等规模的企业提供了一个观察样本:当企业规模达到一定量级(如累计出货100万㎡),当全球环境增长放缓,如何通过组织架构与品牌架构的调整,来继续保持对细分市场的敏捷反应力。母品牌(深德彩 Dicolor):凭借覆盖舞美租赁、创意显示屏、专业地砖屏及高端XR/VP屏的完整产品矩阵,以及继续承担全球供应链、底层技术研发(如260+专利)、大规模制造及租赁基本盘的职能,保持规模效应。并作为技术中台为子品牌输血,保持规模效应带来的成本优势。子品牌(SkyFlex):则化身“特种部队”,专注于高端固装领域,产品线全面覆盖LED一体机、室内固装及户外固装,提供针对性解决方案;并通过独立的品牌形象、差异化的产品逻辑(如主打TCO优势而非低价)以及专业化的服务体系,垂直穿透特定场景。这种“母体平台化 + 子品牌垂直化”的矩阵模式,本质上是对产业分化趋势的一种理性回应。对于整个产业而言,深德彩的探索或许也提出了一个新的命题:未来的核心竞争力,是否不再取决于谁能生产更多的屏幕,而在于谁能在特定的细分场景中,提供更懂客户的技术解法与商业模型。行家说Research总体来看,深德彩推出SkyFlex,意味着其正式构建了“舞台租赁 + COB专业显示 + 商业显示 + 高端固装”的多品牌、多赛道业务矩阵。 当然,这一模式能否最终跑通,不仅取决于战略设计的精巧度,更取决于未来几个关键项目中的实际交付表现,以及SkyFlex能否真正在用户心智中建立起区别于通用品牌的壁垒。这场从“做大”到“做深”的转型之旅,才刚刚启程。但它的航程与成效,值得整个产业持续关注。END相关阅读:直击ISLE:10+LED显示屏厂亮点汇总行业共识:COB与MIP进入差异化共存阶段您的点赞是我们进步的动力!