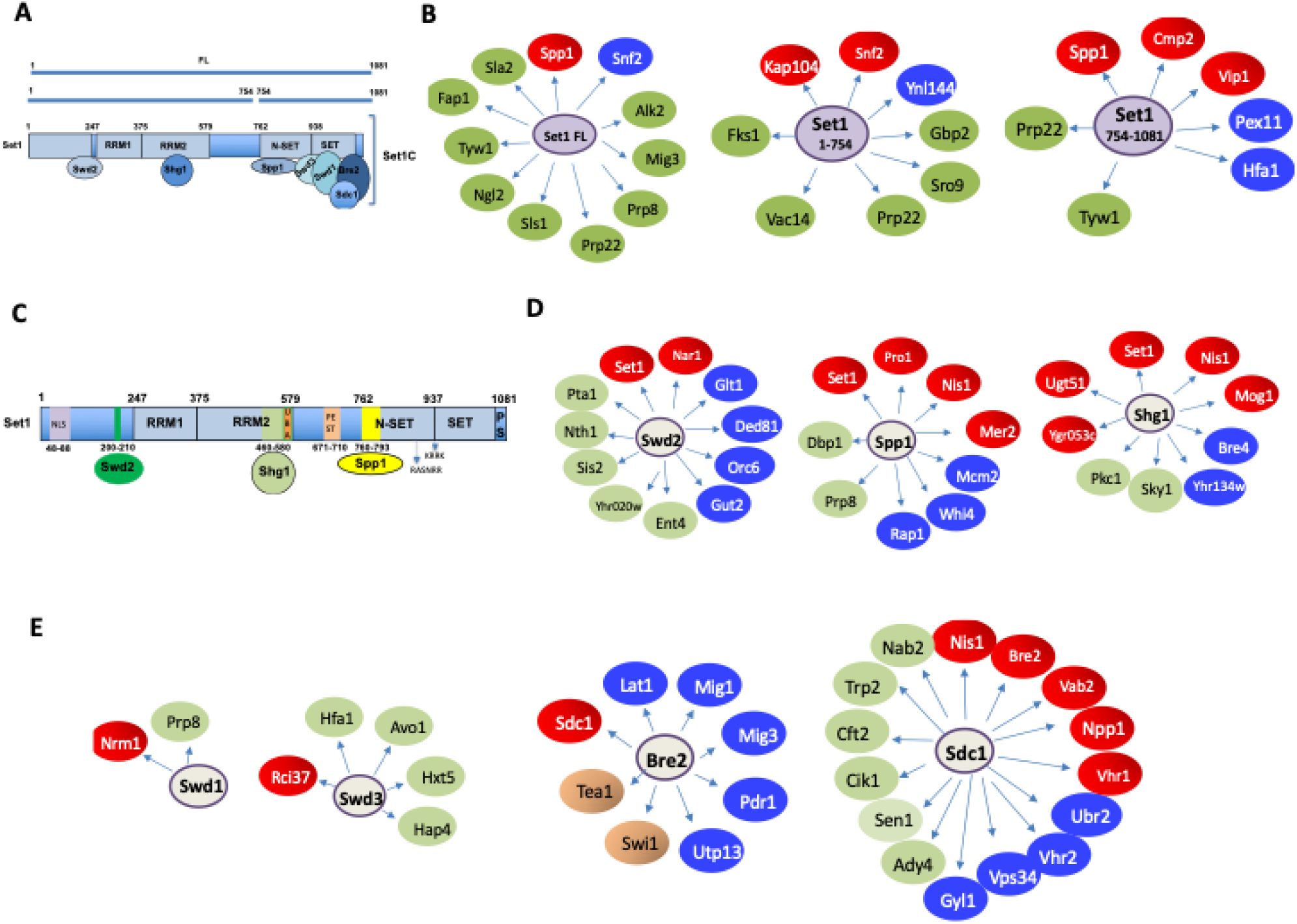
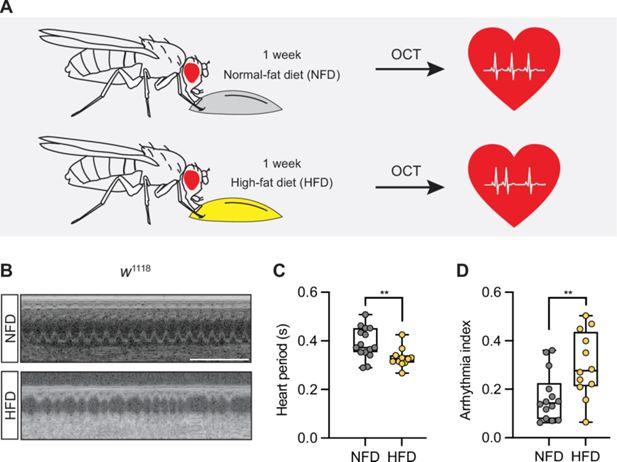
Systematic yeast two-hybrid screening identifies novel functions for SET1C/COMPASS
AbstractSet1 is the catalytic subunit of SET1C or COMPASS, which methylates histone H3K4 and serves as a scaffold for the association of seven tightly bound polypeptides. We have employed yeast two-hybrid screenings to determine the interactome of Set1 and each subunit, providing a unique resource for exploring known and novel roles of the complex. Our screenings identified a multitude of interactors involved in chromatin regulation, DNA replication, meiotic breaks, and Ty transposition, processes previously associated with SET1C. Consistent with Set1 being an RNA-binding protein, the screens link SET1C to multiple aspects of RNA biogenesis, including pre-mRNA splicing and polyadenylation. The results reveal that Set1 interacts with several importins and with RGG motif-containing proteins, providing insights into the mechanisms by which Set1 moves between cytoplasmic and nuclear compartments. We demonstrate that the transcriptional corepressor Nrm1 is methylated by SET1C in vitro suggesting that H3K4-like domains may represent a class of non-histone substrates for SET1C. We further reveal that reconstituted SET1C interacts with the AT hook domain of the chromatin remodeler Snf2 and methylates multiple arginines within this domain. In vivo, we report that the ARTSTRGR AT-hook motif is methylated in a Set1-dependent manner revealing new interplay between lysine and arginine methylation.IntroductionPost-translational modifications of histones shape the chromatin landscape and provide essential mechanisms of regulating DNA accessibility, thereby controlling gene expression, genome maintenance and transmission. Multiprotein complexes of the SET/MLL family methylate histone H3 on lysine 4 (H3K4), which modulates multiple aspects of genome biology (Ruthenburg et al, 2007). Each complex contains additional factors specifying their recruitment to explicit chromatin domains and their specific biological effects (Cenik & Shilatifard, 2021). The budding yeast Set1 complex called COMPASS (for Complex of Proteins Associated with Set1) or SET1C has proved to be an excellent model to study the SET1/MLL family complexes. In Saccharomyces cerevisiae, all H3K4 methylation is carried out by SET1C that is composed of Set1, the catalytic subunit, acting as a scaffold for seven other components (Swd1 [RbBP5], Swd2 [WDR82], Swd3 [WDR5], Bre2 [ASHL2], Sdc1 [DPY30], Spp1 [CFP1] and Shg1 [BOD1]) (Nagy et al, 2002; Briggs et al, 2001; Roguev et al, 2001; Miller et al, 2001). Swd1, Swd3, Bre2 and Sdc1 copurify with the SET domain of Set1, yet none of these proteins can interact alone with the SET domain, suggesting that interactions between subunits are required for catalytic domain formation (Kim et al, 2013). The crystal structure of the SET domain of Set1 associated with Swd1, Swd3, Bre2, Sdc1 shows that the catalytic module is organized by Swd1, whose C-terminal tail nucleates Swd3 and a sub-complex formed by Bre2-Sdc1 (Qu et al, 2018; Hsu et al, 2018). For its side, Spp1 associates with the N-SET domain of Set1, while Shg1 binds to the central region of Set1 and Swd2 contacts the N-terminal region of Set1 (Kim et al, 2013; Roguev et al, 2001; Dehe et al, 2006; Halbach et al, 2009). Swd1 contacts Spp1 and also interacts with the N-terminal region of Set1, suggesting that the C- and N-terminal regions of Set1 may interact with each other (Qu et al, 2018; Acquaviva et al, 2013b; Jeon et al, 2018). Such SET1C organization was confirmed by cryo-electron microscopy and cross-linking experiments performed on the full-length complex (Wang et al, 2018). Interestingly, SET1C has a remarkable mode of assembly that is initiated in the cytoplasm while the nascent Set1 polypeptide emerges from the ribosome. Set1 is initially bound during its translation, by Shg1, Spp1 and Swd1, then Swd2, Swd3, Bre2 and Sdc1 associate with the initial pre-complex to form the full SET1C. This explains why Set1 is associated with its own mRNA (Luciano et al, 2017; Halbach et al, 2009).Swd2, the only essential SET1C subunit, is also part of the APT complex (for Pta1-associated), a subcomplex of the Cleavage Polyadenylation Factor (CPF) involved in the transcriptional termination of mRNA and snoRNA suggesting a functional link between SET1C and 3’ end formation/termination (Cheng et al, 2004; Nedea et al, 2003; Dichtl, 2004). Set1 has also been linked genetically to the Nrd1-Nab3-Sen1 (NNS) complex (Kim et al, 2006; Arigo et al, 2006), but intriguingly, it was reported to have two seemingly contradictory effects on ncRNA termination: on the one hand Set1 was suggested to promote efficient snoRNA termination mediated by Nrd1 (Terzi et al, 2011), and on the other hand it appeared to interfere with the early termination of a large number of ncRNAs (Castelnuovo et al, 2014; Margaritis et al, 2012). Interestingly, various lysines distributed between Nrd1, Nab3 and Sen1, are methylated, notably lysine 363 in the RNA Recognition Motif (RRM) of Nab3 whose mono methylation depends on Set1 and Set3 (Lee et al, 2020).Set1 interacts co-transcriptionally with the RNA polymerase II Carboxy Terminal Domain (PolII CTD) phosphorylated on Ser5 of the heptad repeats, producing an H3K4 methylation gradient that starts at nucleosome +1 and fades away from the promoter (Soares et al, 2017; Ng et al, 2003). It was recently shown that the N-terminal region of Set1 and Swd2 interact cooperatively with the CTD (Carboxy Terminal Domain) of Rbp1 (RNA polymerase II large subunit) promoting SET1C recruitment to transcription elongation complexes at the 5′ ends of genes (Bae et al, 2020). Deletion of residues 200-210 of Set1 abolishes interaction with the Rpb1-CTD and Swd2, while deletion of the first 200 amino acids of Set1 strongly reduces trimethylation of H3K4 (H3K4me3) at the 5’ end of transcribed genes (Bae et al, 2020). Interestingly, Swd2 is ubiquitinated by Rad6/Bre1 and preventing Swd2 ubiquitination affects H3K4 trimethylation with a concomitant reduction in Spp1 recruitment to chromatin suggesting a cross-talk between Swd2 and Spp1(Vitaliano-Prunier et al, 2008). In a similar vein, di- and tri-methylation of H3K4 by Set1, which depends on prior ubiquitination of histone H2B by Rad6/Bre1 (Dover et al, 2002; Sun & Allis, 2002), requires a contact between Spp1 and Swd1 (Hsu et al, 2019; Jeon et al, 2018). Other modes of SET1C recruitment to chromatin must exist in the absence of the Set1 N-terminal region, probably via direct interactions between SET1C’s catalytic domain and the nucleosome (Jeon et al, 2018; Dehé & Géli, 2006; Thornton et al, 2014).The central region of Set1 contains two RRMs positioned in tandem (Trésaugues et al, 2006) through which Set1 binds directly to RNA. This binding contributes to retain Set1 in the 5’ region of genes thus facilitating their H3K4 trimethylation (Luciano et al, 2017; Battaglia et al, 2017; Sayou et al, 2017). Both the N-SET domain and Spp1 also contribute to Set1 binding to RNA. Set1 associates post-transcriptionally with transcripts produced by specific classes of genes including snRNA, Ty1 and a number of genes involved in adaptive responses (Luciano et al, 2017). The dRRM itself is flanked by an autoinhibitory region that negatively regulates H3K4me3 (Schlichter & Cairns, 2005). It is possible that in the context of full-length Set1, the alternative mode of SET1C recruitment may be inhibited by the autoinhibitory domain.In S. cerevisiae, H3K4me3 methylation is counteracted by the demethylase Jhd2, a conserved JARID1 family protein (Huang et al, 2010; Liang et al, 2007). The erasure of ancestral histone methylation states results from both active enzymatic demethylation by Jhd2 and passive dilution of parental histones during replication (Radman-Livaja et al, 2010). SET1C is a dimer and its dimerization depends on the Sdc1 subunit (Choudhury et al, 2019). It has been proposed that the symmetrical methylation of H3K4 on nucleosomes is a consequence of the dimeric nature of SET1C and that Jhd2 preferentially demethylates asymmetrical H3K4me3 (Choudhury et al, 2019). Interestingly, H3K4me3 at environmental stress genes depends on H3-P16 isomerisation, a process that controls K4me3 by balancing the actions of Jhd2 and Spp1 (Howe et al, 2014).The complexity of the S. cerevisiae SET1C, the specific arrangement of subunits within the complex, and the specific roles of certain subunits, notably Spp1 and Swd2 (Acquaviva et al, 2013a), raise the question of the contribution of SET1C and of its subunits to multiple processes. Overall, H3K4 methylation, SET1C and its individual subunits have been involved in multiple processes in S. cerevisiae such as meiotic recombination (He et al, 2019; Sollier et al, 2004; Borde et al, 2008; Acquaviva et al, 2013b; Sommermeyer et al, 2013; Karányi et al, 2018; Adam et al, 2018), stress response and epigenetic transcriptional memory (D’Urso et al, 2016; Kim & Buratowski, 2009; Weiner et al, 2012), DNA repair (Faucher & Wellinger, 2010), telomere and rDNA silencing (Jezek et al, 2023; Corda et al, 1999; Briggs et al, 2001; Bryk et al, 2002; Santos-Rosa et al, 2004; Nislow et al, 1997), cell wall biogenesis (Nislow et al, 1997), chromosome segregation (Beilharz et al, 2017; Zhang et al, 2000), antisense transcription (Murray et al, 2015; van Dijk et al, 2011; Margaritis et al, 2012; Castelnuovo et al, 2014), transcription termination (Kaczmarek Michaels et al, 2020; Nedea et al, 2003; Dichtl, 2004; Cheng et al, 2004; Nedea et al, 2008; Terzi et al, 2011; Soares & Buratowski, 2012; Castelnuovo et al, 2013; Lee & Wang, 2018), Ty silencing (Luciano et al, 2017; Berretta et al, 2008), chronological aging (Gong et al, 2023; Walter et al, 2014; Mei et al, 2019), ergosterol homeostasis (South et al, 2013), stress response (Deshpande et al, 2022; Nadal-Ribelles et al, 2015), lipid homeostasis (Giaever et al, 2019), and DNA replication (Ghaddar et al, 2023; Sollier et al, 2004; Rizzardi et al, 2012; Chong et al, 2020; Delamarre et al, 2020; Santos-Rosa et al, 2021; de La Roche Saint-André & Géli, 2021; Serra-Cardona et al, 2022).The multiple roles of Set1 and its subunits led us to perform global two-hybrid screening to identify interactors either of Set1-full length, or of its N- and C-terminal regions and of each of the individual subunits (Swd2, Shg1, Spp1, Swd1, Swd2, Sdc1, Bre2). The identification of interactors is discussed not only for each bait, but also as a whole, revealing new potential functions for the complex. In addition, we demonstrate that the transcriptional corepressor Nrm1 and the Snf2 AT-hook are both methylated by SET1C in vitro. In vivo, deleting SET1 abrogates arginine methylation within the ARTSTRGR motif of Snf2 AT-hook. These results suggest that H3K4-like sequences may represent a class of non-histone substrates for SET1C and reveal new interplay between Lysine and arginine methylation. This work is an invaluable resource for further exploring known and unsuspected roles of the SET1C complex and its subunits.ResultsIdentification of Set1 and Set1 subunits interactorsWe produced a total of ten yeast two-hybrid (Y2H) screens (Hybrigenics, See Methods). For the Set1 protein, we performed a total of three screens using as bait the full-length Set1 protein (Set1 FL), the amino-terminal region including amino acids 1-754 or a C-terminal fragment including amino acids 754-1081 (Fig. 1A). We have chosen to separate Set1 into these 2 regions because they have been described as having well-defined properties. The Set1 1-754 fragment includes the domain involved in Set1 recruitment to chromatin, the double RRM (Trésaugues et al, 2006), and the central self-inhibitory domain (Schlichter & Cairns, 2005) that all have regulatory roles (Dehé & Géli, 2006). The Set1 region 754-1081 contains the N-SET domain, the SET domain and the post-SET domain: the latter is capable of methylating H3K4 on its own (Thornton et al, 2014) (Fig. S1). For each of the subunits (SU) of the complex whose organization is described in Fig. 1A we used the whole protein as bait. Each gene encoding Set1 (and its fragments) or its subunits was cloned downstream the Gal4-BD except for Swd2 that was placed upstream the Gal4-BD. These screens have proven their power and effectiveness. In particular, they identified Mer2 as an interactor of Spp1 (Acquaviva et al, 2013b), and the CTD of Rpb1 as an interactor of the N-terminal region of Set1 (Bae et al, 2020) (Fig. S1). The results of the ten Y2H screens are presented in Fig. 1 and Table S2. Each interactor is characterised with a confidence score based on several criteria, including the frequency with which a prey protein is found for a particular screen and the presence of overlapping fragments, which allow the delineation of the interaction domain involved (see Methods). Very high confidence interactors (indicated by a color code) are likely to interact directly with their bait. For Set1 and its N- and C-terminal fragments, the high confidence Y2H interactors are shown in Fig. 1B. All interactors are shown in Table S2. The predicted protein-protein interactions within the Set1 interactors reveal functional information that is detailed in the following paragraphs. The high confident interactors of the seven SET1C subunits are shown in Fig. 1C-E. We found that Spp1, Shg1 and Swd2 interact alone with Set1 (Fig. 1C). The minimum Set1 region for which an interaction is found for each of these 3 subunits is shown in Fig. 1C. The high confidence interactors of the seven SET1C subunits are shown in Fig. 1C-E. We found that Spp1, Shg1 and Swd2 display Y2H interactions with Set1 (Fig. 1C). The high confidence interactors of Spp1, Shg1 and Swd2 are indicated in Fig. 1D (see also Table S2). In contrast, we did not find any interaction between Swd1, Swd3, Bre2 and Sdc1 (SET-c components) and Set1 in the various Y2H screens, suggesting that these subunits must cooperate to bind to Set1. Only Bre2 was found to interact with Sdc1 in both Sdc1 and Bre2 Y2H screens (Fig. 1E). These results are consistent with the structure of the extended SET1C catalytic module and full-length SET1C (Wang et al, 2018; Hsu et al, 2018; Qu et al, 2018).Figure 1.Schematic representation of the Set1 and subunit major interactors identified in the systematic yeast two-hybrid screens.A) Schematic representation of Set1 FL and Set1 fragments 1-754 and 754-1081. B) Set1 FL, 1-754 and 754-1081 major Y2H interactors). C) Interacting regions of Set1 with Swd2, Shg1 and Spp1. D) Swd2, Spp1, Shg1 major Y2H interactors. E) Swd1, Swd3, Bre2, Sdc1 major Y2H interactors. The term “interactor” is used to mean a high confidence two-hybrid interaction, with the limitations that this entails. The color reflects the Predicted Biological Score (see METHODS). Red, highest confidence; Blue, high confidence; Green, good confidence.Set1 1-754 interacts with the importin Kap104We found that Set1 1-754 interacted with very high confidence with the importin Kap104, suggesting a direct interaction (Fig. 2A). Kap104 has been reported to recognize specific cargos containing a specific class of NLS termed PY-NLS (Soniat et al, 2013; Xu et al, 2010). This PY-NLS includes a N-terminal or central hydrophobic or basic motif, which often contains hydrophobic (R/H/K) residues and which can include RGG repeats (arginine-glycine-glycine motifs) and a C-terminal Proline-Tyrosine (PY) dipeptide near the C-terminus. Such cargos include the mRNA export factor Nab2, the subunit of the THO/TREX complex Hrp1, and the transcription factor Tfg2 (Lee & Aitchison, 1999; Süel et al, 2008). Interestingly, Nab2 is a confident interactor of Sdc1 (Fig. 2A). The Set1 interacting domain (SID) of Kap104 extends from residue 359 to 621 and includes the HEAT-like repeat (Yoshimura & Hirano, 2016) (Fig. 2B). Consistent with this result, we identified two PY-NLS in the N-terminal region of Set1 that are likely recognized by Kap104 (Fig. 2C). Along the same line, Shg1, which binds RRM2, interacted with very high confidence with Mog1 (Oliete-Calvo et al, 2018). Mog1 has been involved in the modulation of the nucleotide state of Ran-GTP in the nucleus and of Ran-GDP in the cytoplasm, thereby conferring directionality to the nuclear import pathway (Baker et al, 2001). Mog1 has been reported to interact directly with the Ran homologue Gsp1 (Oki & Nishimoto, 1998) (Fig. 2A). We have also identified, Kap123, Msn5 (Kap142), Nup82 and Mtr10 as interactors of Set1 1-754 that are also involved in protein import/export (Fig. 2A). Kap123 is a major karyopherin that recognizes NLS of cytoplasmic H3 and H4 (An et al, 2017) while Msn5/Kap142 was shown to mediate the import into the nucleus of the subunits of RPA (Yoshida & Blobel, 2001). Nup82 for its part belongs to a module at the cytoplasmic face of the NPC and interacts with karyopherins (Beck & Hurt, 2017) (Fig. 2A). Finally, Mtr10 has been implicated in the nuclear import of the mRNA-binding protein Npl3 (Senger et al, 1998; Pemberton et al, 1997). Collectively, these multiple interactions reveal insights for understanding the nuclear import of Set1, which was reported to bind co-translationally with Shg1, Swd1, and Spp1 (Halbach et al, 2009).Figure 2.Set1 1-754 interacts with RGG proteins and the importin Kap104.A) RGG proteins and import/export proteins interacting with Set1 1-754, Set1 754-1081 and Spp1, Shg1, and Sdc1. B) Set1 interacting domain (SID) (blue) within Kap104. Heat like repeat 9 and 10 are represented in purple C) PY-NLS in the N-terminal region of Set1. D) SID (blue) and RGG motif (green) within the RGG proteins. The interaction domains indicated represent the minimal overlapping DNA sequence present in multiple independent Y2H interacting clones of the same gene. Each genomic fragment of a Y2H clone was analyzed, and the shared overlapping region for given gene was determined to be the only common element among all interacting clones. As such, this region represents the minimal sequence required for interaction.Set1 1-754 interacts with Snf2 and RGG motif proteinsSet1 1-754 and Set1 FL interacted with very high confidence with Snf2, the catalytic subunit of the SWI/SNF chromatin remodeling complex (Côté et al, 1994) (Fig. 1B). Interestingly, two additional Snf2 complex components, Snf5 and Swi1, have been identified in the Set1FL screen (Table S2). Together these observations strongly support the idea that the suggested interaction of Set1 and SWI/SNF is of biological significance (Hirschhorn et al, 1992). The various Y2H screens revealed Set1 and its subunits interacted with a number of proteins involved in chromatin structure regulation although the relevance of these interactions remains to be demonstrated (Fig. 3). Interestingly, the SID region of Snf2 is juxtaposed to an RGG motif composed of several RGG/RG repeats that is often found in RNA-binding proteins (Thandapani et al, 2013) and may be the substrate for arginine methyltranferases (McBride et al, 2005) (Fig. 2A, 2D). RGG repeats are multifunctional motifs that mediate RNA and DNA binding and nuclear import. They have been involved in RNA metabolism and chromatin dynamics possibly via arginine methylation, which modulates RNA affinity and nuclear localization (108). Along the same line, Set1 1-754 also interacted with the mRNA export factor Gbp2 (Poornima et al, 2021) and the nucleolar protein Nop1, both of which contain an RGG motif (Fig. 2A, 2D). Other RGG proteins such as polyadenylated RNA-binding protein Nab2 and the RNA-helicase Dbp1 interacted with Set1 754-1081 (and Sdc1) and Spp1, respectively (Fig. 2A, 2D). For each of these proteins, the putative SID includes the RGG motif. Of note, both Nop1 and Nab2 are methylated by the Arginine methyltransferase Hmt1 (Smith et al, 2020; Green et al, 2002).Figure 3.Chromatin regulators identified in all the two-hybrid screens.The lines indicate the individual proteins involved in the Y2H interaction. Red line refers to a very high confidence Y2H interaction. All Y2H interactors are described in Table S2. Interactors are grouped according to the complex to which they belong.Our screens indicate that the minimal region of Snf2 interacting with Set1 1-754 was located between Snf2 residues 1430-1499. We further refined this interaction by Y2H testing of five Set1 fragments covering the entire Set1 length against the Snf2 1349-1650 region. The two-hybrid analysis revealed that Snf2 (1349-1650) interacted with the N-terminal domain (Set1 1-230) and the N-SET region (Set1 762-938) (Fig. S2). To validate these interactions we obtained recombinant protein where GST was fused to the C-terminal domain of Snf2 (Snf2C), the AT-hook (Snf2C-AT-hook) and the Bromo domain (Snf2C-Bromo) (Kim et al, 2010) (Fig. 4A). We performed pull-down assays to test the interaction of the GST-fusion proteins with reconstituted SET1C or with the SET1C762 complex in which Set1 has a N-terminal truncation of the first 761 residues, respectively (See METHODS) (Fig. S3 A-E). We found that SET1C as well as the SET1C762 complex were both associated with Snf2C and Snf2C-AT-hook but not to Snf2C-Bromo (Fig. 4 B, C), indicating that AT-hook is a key region for interaction with SET1C. We then delineated the minimal AT-hook region required for interaction with SET1C and found that residues 1461-1547 are essential for binding to SET1C (Fig. 4 D-E). Collectively, these results indicate that SET1C interacts with Snf2C-AT-hook region suggesting that the Set1 and Snf2 complexes cooperate to modify chromatin structure.Figure 4.SET1C interacts in vitro with Snf2C-AT-hook.A) A schematic diagram depicting the domains of Snf2 and the Snf2 fragments used in this study, along with the SDS-PAGE/Coomassie staining of the purified GST-tagged Snf2 fragments. B and C) GST pull-down assay using purified GST-tagged Snf2 fragments. The purified SET1C (B) or SET1C-C762 complex (C) was mixed with GST-tagged Snf2 fragments, followed by GST pull-down, and the bound proteins were analyzed by immunoblotting. D) A schematic diagram illustrating Snf2 fragments with a more detailed breakdown of the AT-hook domain, along with the SDS-PAGE/Coomassie Blue staining of the purified Snf2 fragments. The lysines present in the AT-hook are represented by the letter K. E) GST pull-down assay using purified GST-tagged Snf2 fragments and SET1C.SET1C is involved in multiple aspects of RNA biogenesisSet1 was previously shown to bind RNA nascent transcripts through its dRRM contributing to position Set1 and H3K4me3 predominantly to the 5′ regions of genes. Of note, Set1 showed a higher occupancy within introns, at transcripts from ribosomal DNA (rDNA), and tRNAs (Luciano et al, 2017; Battaglia et al, 2017; Sayou et al, 2017). Moreover, Set1 also binds post-transcriptionally to Ty1 retrotransposon transcripts and mRNA encoding specific transcription factors genes (Luciano et al, 2017). The SET1C Y2H interactome revealed that many proteins involved in RNA biogenesis were interacting directly or indirectly with Set1 or its subunits (Fig. 5). We find a strong connection between SET1C and pre-mRNA processing, as highlighted by the identification of multiple spliceosome subunits (Fig. 5). Some spliceosome subunits were found in several screens (Fig. 1B, D; Table S2). For instance, the Prp8 subunit of the U5 snRNP that function in critical molecular rearrangements during the splicing process (Grainger & Beggs, 2005) interacted with Set1 FL, Spp1, and Swd1. Similarly, Prp22 interacts with Set1FL and Set1 754-1080. Prp22 is a DEAH-box helicase that associates with newly spliced mRNA and promotes its release from the spliceosome (Will & Lührmann, 2011). We have refined the interaction region between Set1, Prp8 and Prp22, showing that Prp8 and Prp22 interact strongly with Set1-F4. (n-SET). Prp22 interacts in addition with Set1-F1 (Fig. S2). We have further strengthened the link between Set1 and Prp22 by showing that Set1 is co-immunoprecipitated with Prp22 in vivo in an RNA-independent manner. (Fig S4A, B). The preferential binding of Set1 to genes with introns (Luciano et al, 2017) and its interaction with splicing factors, in particular Prp22, suggest that Set1 may be involved in late splicing events. Alternatively, Prp22 and/or other splicing factors could be involved in H3K4 methylation.Figure 5.The SET1C Y2H interactome identifies proteins involved in RNA biogenesis.All Y2H interactors are described in Table S2. The processes linked to RNA metabolism in which the different interactors are involved are shown in the figure. The green lines linking Prp22 to Set1FL/Set1 754-1080 and Prp8 to Set1 FL and Spp1 indicate interactions with a high degree of confidence.We also found a number of factors involved in rRNA processing that could be related to the binding of Set1 to ncRNA transcripts derived from the rDNA intergenic spacer regions (Sayou et al, 2017) (Fig. 5). Next, and consistent with the observation that SET1C regulates the choice of the polyadenylation site and the recruitment of the cleavage/polyadenylation complex (Kaczmarek Michaels et al, 2020), the Y2H screens revealed multiple potential interactions between SET1C and factors involved in RNA polyadenylation (Fig. 5). Some of these interactions are known, such as the interactions between Swd2 with Pta1 and Ref2, all three proteins belonging to the APT complex (Nedea et al, 2008). Interestingly, we found that Set1 1-754 interacts with Not1 and Not4 that belongs to the ubiquitin-protein ligase CCR4-NOT (Liu et al, 2001) (Fig. 5). This complex was shown to be involved in the regulation of H3K4me3 via a ubiquitin-dependent pathway (Mulder et al, 2007; Laribee et al, 2007) that was subsequently linked to Jhd2 degradation (Huang et al, 2010; Mersman et al, 2009). These results raise the question of which of the Y2H interactions described in this study are linked to the regulation of H3K4 methylation states.Finaly and somewhat surprisingly, we found that many Y2H interactors of Set1 1-754 were involved in tRNA nuclear transport, modification, and synthesis (Fig. 5). Swd2, which interacts with the Set1 N-terminus, exhibit a 2H interaction with a high degree of confidence with the cytoplasmic asparaginyl-tRNA synthetase Ded81 and the prolyl-tRNA synthetase Yhr020 while Sdc1interacted with the glycyl-tRNA synthase Grs1 and Bre2 with the glutamine tRNA synthetase Gln4. Of note, Trm1 (Liu et al, 1998) and Trm732 (Guy et al, 2012) are both directly or indirectly involved in tRNA methylation and form part of what has been defined as the cell’s global methytransferome (Giaever et al, 2019).SET1C Y2H interactors are involved in several aspects of DNA transactionsAs mentioned in the introduction, it has been shown that SET1C, and in particular Spp1, regulate the selection of meiotic breaks, the progression of the replication fork, DNA repair, chromosome segregation, and transposition of Ty elements (Deshpande & Bryk, 2023). We have classified all Set1 and subunit interactors according to these SET1C roles (Fig. S5). We found a number of interactors involved in meiosis. In particular, Spp1 not only interacts with Mer2, but also with the meiosis specific DNA helicase Mer3 (Nakagawa & Kolodner, 2002) that interacts also with Swd1 (Fig. S5). We further found Set1 interactions with the kinetochore proteins Spc25 and Cbf2 suggesting that Set1 could be transiently localized at the spindle pole body. Regarding the biology of retrotransposons, we previously reported that Set1 binds post-transcriptionally to Ty1 mRNA and represses Ty1 mobility (Luciano et al, 2017). Interestingly, the two-hybrid screens reveal that Set1 1-754 interacted with Gag capsid-like proteins of Ty1 (Fig. S5) raising the possibility that Set1 binding to Ty1 mRNA is linked to the interaction of Set1 1-754 with Gag. Concerning the role of Set1 and subunits in DNA replication and repair, we report multiple potential interactions (Fig. S5). In particular, Swd2, Swd1, and Spp1interacted with high confidence with Orc6, Nrm1, and Mcm2, respectively (Fig. 1 and Fig. S5). Orc2 was previously described to interact physically with Spp1 (Kan et al, 2008). We recently reported that Spp1 is recruited at replication forks stalled at the Tus/Ter barrier independently of its interaction with Set1 (Ghaddar et al, 2023). Interestingly, in the Y2H screen Spp1 interacted with the extreme C-terminal region of Mcm2 (791-867) (Fig. S6A) that corresponds to a non-conserved accessible alpha-helix within the MCM complex (Li et al, 2015b). We fused Spp1 and Mcm2 to GST and performed GST pull-down experiments. We confirmed in vitro in both directions a weak interaction between Spp1 and Mcm2 (Fig. S6B). Whether Spp1 is recruited by Mcm2 at stalled replication fork remains to be determined. On its side Nrm1 inactivates MBF, a major regulator of the G1/S transcription (de Bruin et al, 2006). During replication stress, Nrm1 phosphorylation by the checkpoint kinase prevents its binding to MBF target promoters leading to the activation of G1/S transcription (Travesa et al, 2012). An exciting idea is that Swd1 recruits Nrm1 to stalled forks by promoting its phosphorylation by Rad53. Swd1 would play a role in linking replication stress and transcriptional regulation via Nrm1. Of note, Nrm1 was identified as a gene required for the cell-cycle pattern of H3K79me2 during early S phase (Schulze et al, 2009). The interaction between Swd1 and Nrm1 is described in more detail below.Finally, Y2H screening indicated that Set1 and its subunits interacted with a number of proteins involved in protein SUMOylation (Fig. S5). The proteins were either involved in SUMO conjugation or SUMO-dependent degradation. Remarkably, the C-terminus of Nis1 (360-407) that contains a potential SUMO-binding site (Hannich et al, 2005) was identified a high-confidence interactor of Spp1, Shg1, and Sdc1 (Fig. 1). Nis1 is localized at the bud neck (Iwase & Toh-e, 2001) at the vicinity of the septin collar containing several highly SUMOylated proteins (Shs1, Cdc11) (Wykoff & O’Shea, 2005) and has been implicated in preventing bud recovery at the site of division (Meitinger et al, 2014). We thus sought to confirm biochemically the interaction of Nis1 with the three SET1C subunits. We fused the GST to Spp1, Sdc1, and Sgh1 to perform pull-down experiments with in vitro translated Nis1. We confirmed the interaction of Nis1 with Spp1, and Sdc1, but not with Sgh1 (Fig. S7). However, mass spectrometry analyses on TAP-Nis1 did not reveal the presence of SET1C subunits (Table S3) suggesting that interaction between Nis1 and Spp1/Sdc1 might be transient. The relevance of the Nis1 and other putative interactors remains unclear, especially as many of those proteins are not known to be located in the nucleus. Interestingly, Nis1 has been reported to shuttle from the bud neck to the nucleus when overexpressed (Perez & Thorner, 2019). In Fig. S8, we have identified a number of interactors, including Sdc1 and Spp1, showing changes in localization following hypoxia (Henke et al, 2011). Of note, one of the two human SET1C homolog SET1B has a cytoplasmic location with functions unrelated to H3K4 methylation (Wang et al, 2017). Combined, these observations raise the question of the minor or transient localization of Set1 and its subunits outside the nucleus, or conversely of the transient localization of interactors within the nucleus.Set1 is SUMOylatedGlobal analyses of SUMOylated proteins in fission yeast revealed that Set1 and Spf1 (Spp1) were SUMOylated (Shin et al, 2005; Nie et al, 2015). As Set1 had two-hybrid interactions with Slx5 and Wss1, both of which act on SUMOylated proteins as they undergo protein degradation (Mullen et al, 2010), we tested whether Set1 can be SUMOylated. Cells expressing Myc-Set1 (Dehe et al, 2006) or transformed with pPB66-SET1 expressing GBD-Set1-FL (GAL4 binding domain) were transformed with a plasmid encoding His6-SUMO or a control plasmid. Proteins were purified on Ni-NTA agarose beads and detected by Western blot either using anti-MYC or anti-GAL4 antibodies. The results revealed that Set1 can be mono-SUMOylated (Fig. 6A) or di-SUMOylated (Fig. 6B). This difference may be due to the fact that GBD-SET1-FL is under the control of the ADH promoter in plasmid pGB66 and thus overexpressed.Figure 6.Set1 is SUMOylated.6His-SUMO–conjugated proteins were purified from cells transformed (+) or not transformed (−) with a plasmid encoding 6His-SUMO under control of the CUP1 promoter. Cell lysates (Input) and Ni-purified material (Elutes) were analyzed by Western blotting with an anti-MYC antibody (A) or and anti-GAL4 antibody (B-D). Analysis of 6His-SUMO -conjugated forms of (A) genomically MYC-tagged Set1 or (B) GB-Set1 transformed cells was performed (left panels), in both the cases SUMO expression and efficiency of purification were controlled using an anti-SUMO antibody (right panels). C) SUMOylation analysis of Set1 fragment F3+F4 (aa. 351-956) WT and the K769R mutant. D) SUMOylation analysis of Set1 fragment F5 (aa. 956-1080) WT, single mutants K1055R and K1060R, and the double mutant K1055R/K1060R mutant.We sought to refine the SUMOylated Set1 region. We transformed the plasmid encoding His6-SUMO and the control plasmid into cells expressing Set1 fragments F1, F2, F3, F4, F3+F4, and F5 (Fig. S9A). We expressed F3+F4 instead of the isolated F3 and F4 fragments in order to preserve the K769-centered motif predicted to be highly SUMOylated (Fig. S9B). F1, F2, F3, and F4 were not SUMOylated (not shown) while the F3+F4 fragment was clearly SUMOylated (Fig. 6C). In order to identify whether K769 is important for F3+F4 SUMOylation, we mutated K769 to R. We show that introduction of K769R into F3+F4 abolishes its SUMOylation (Fig. 6C), indicating that K769 is likely to be the SUMOylated lysine within the F3+F4 fragment. We then found that F5 was also SUMOylated (Fig. 6D). In this case, the substitution K1055R, K1060R, or the double substitution, does not affect the SUMOylation of F5 indicating that this motif also predicted to be SUMOylated with a high score is not the SUMOylated motif (Fig. 6D). Taken together, these experiments indicate that Set1 can be SUMOylated in the N-SET and SET domains, in the interaction region of Spp1 and Swd1-Swd2-Bre2-Sdc1, respectively. This raises the possibility that SUMOylation regulates the interaction of Set1 with its subunits, in particular with Spp1, which interacts dynamically with SET1C (Ghaddar et al, 2023; D’Urso et al, 2016; Karányi et al, 2018; Serra-Cardona et al, 2022). Interestingly, in mammals, the SUMO peptidase SENP3 (the ortholog of Ulp1 in budding yeast) interacts with MLL1 and MLL2, catalyzing the deSUMOylation of RbBP5 (Swd1). This process regulates the association of specific subunits of MLL1/MLL2, such as menin and Ash2L (Bre2), with the DLX3 gene, which plays a role in osteogenic differentiation (Nayak et al, 2014).The transcriptional corepressor Nrm1 interacts with SET1CScreening with SWD1 yielded a total of forty clones, fourteen of which contained fragments of the NRM1/YNR009w gene, which encodes a basic protein of 249 amino-acids (Fig. 7A). Its amino-terminus carries a putative D-box motif that was found to destabilize the protein (de Bruin et al, 2006). The Y2H results furthermore delineated a central domain (amino-acids 58 to 195) mediating the interaction with Swd1. A sequence motif within this domain displays strong similarity to the amino-terminus of histone H3; lysine 118 within Nrm1 (Nrm1-K118) aligns with lysine 4 of histone H3, the substrate of SET1C. We refer to this sequence as H3K4-like domain. The similarity of the H3K4 like domain and the H3K4 modification site suggested that Nrm1 may represent a non-histone substrate for SET1C. To test this idea, we initially performed in vitro methylation experiments with partially purified SET1C, S-adenosyl (methyl-3H) methionine and peptide substrates. We found that a H3K4 peptide was efficiently methylated, but a peptide carrying a H3K4A substitution was not (Fig. 7B). Also, no methylation was observed with a peptide encompassing K118 of Nrm1. The possibility remained that this peptide lacked parts of Nrm1 that were required for methylation. For this purpose, we obtained recombinant protein where the N-terminus of Nrm1 was fused to the 42 kDa maltose binding protein (MBP). Interestingly, we observed in vitro methylation of this protein by SET1C but not with MBP only (Fig. 7C). Note, however, that histone methylation occurred much more efficiently under these reaction conditions. Next, we asked whether K116 and K118 of Nrm1 were required for the observed methylation activity. Fig. 7D shows that wild-type and mutant MBP-fusion proteins were found to be methylated suggesting that the H3K4-like domain is not required for the observed SET1C methylation activity.Figure 7.Nrm1 is methylated in vitro by SET1C.A) Schematic representation of the S. cerevisiae Nrm1 protein. Nrm1 carries a D-box sequence at its amino-terminus and a central domain that mediated interaction with the Swd1 subunit of SET1C in a yeast two-hybrid screen. A H3K4 like domain that closely resembles the modification site of SET1C in histone H3 is contained within the Swd1 interaction domain. Identical and similar amino-acid positions are shown in italic. Lysine 118 (K118) of Nrm1 aligns with lysine 4 of histone H3; also indicated are K116R and K118A mutant sequences. B) In vitro methylation reactions using partially purified SET1C, 3H-SAM and the indicated peptides. Radioactive reaction products retained on Whatman P81 filter paper following extensive washes were measured by scintillation counting. Each reaction was done in triplicate and error bars indicate the standard deviation. C) In vitro methylation reactions as in (B), however, core histone (2, 4 and 6 mg) and recombinant MBP and MBP-Nrm1 fusion protein (0.5, 1 and 2 mg) were tested as substrates for methylation. Reaction products were separated on 4 to 12% NuPAGE gels and analyzed by fluorography. D) In vitro methylation reactions as in (C), however, core histone (2 mg) and recombinant MBP-Nrm1 wild-type and K118A and K116R fusion protein (0.5, 1 and 2 mg) were tested as substrates for methylation. (upper panel). Note that the molecular mass marker proteins (MM) have been resolved together with the core histone methylation reaction in the same lane. The asterisk indicates migration of a non-specific background signal of unclear origin. The substrate proteins included in the reactions were analyzed on a parallel gel and stained with colloidal Coomassie G-250 (lower panel). E) H3K4-like proteins. Shown are selected proteins containing sequences similar to the modification site found in histone H3. An exhaustive list can be found in Table S4.The H3K4-like domain in Nrm1 raised our attention to other yeast proteins that carry such sequences. We used the scansite search algorithm (http://scansite.mit.edu) to systematically identify sequence motifs that are related to the SET1C modification site in histone H3. Search parameters included four to six identical residues of the ARTKQT sequence that is found at the H3K4 modification site. In addition, we did allow for biochemically equivalent amino acid changes. Fig. 7E shows eight candidate proteins based on their function and the sequence context of the H3K4-like domain. Importantly, some of these gene products are involved in cellular pathways that functionally overlap with SET1C, e.g. transcription (Not5), rDNA silencing (Irs4) and cell-cycle control (Dbf2 and Dbf20), increasing the possibility of occurring physical and functional interactions.SET1C Y2H interactors regulate metabolism and stress responsesSet1 was reported to regulate ergosterol levels (South et al, 2013). We identified Erg9, Ugt51, Vhr1, Vhr2 and Ste20, all involved in ergosterol metabolism (Daicho et al, 2020; Lees et al, 1995; Warnecke & Heinz, 1994). Erg9 and Ste20 interacted with Set1 while Ugt51 and Vhr1/Vhr2 were high confidence interactors of Shg1 and Sdc1, respectively (Fig. S10). Along the same line, the various Y2H screens revealed numerous genes involved in phosphatidyl inositol metabolism interacting with Set1 and its subunits. In particular Vip1 is a high-confidence interactor of Set1 754-1081 (Fig. 1B). Vip1 is a bifunctional inositol pyrophosphate kinase and phosphatase that regulates IP7 levels in the inositol pyrophosphate (PP-IP) synthesis pathway (Lee et al, 2007; Mulugu et al, 2007). Interestingly, Vip1 was reported to regulate the environmental stress response (ESR) through IP7 that activates the HDAC Rdp3 (Worley et al, 2013) suggesting potential new avenues to explain ESR regulation by Set1 (Weiner et al, 2012). Along the same line, Swd1 and Swd3 were identified in a screen aimed to identify genes that negatively regulate the PHO pathway in a Vip1-dependent manner (Choi et al, 2017). How the putative interaction between Set1 754-1081, which also interacts with Pho23 subunit of the Rpd3L complex, and Vip1 fits into these processes remains to be discovered. Of note, Vip1 has been shown in Arabidopsis thaliana to change localization upon hypo-osmotic stress from the cytosol to the nucleus (Takeo & Ito, 2017). On their side, Y2H Interactors identified as stress-responsive genes interact either with Set1 1-754 or Swd2 (Fig. S10). Swd2 interacted with high confidence with Nar1, an essential Fe/S protein required for the assembly of cytosolic Fe/S proteins (Balk et al, 2004) and the calcineurin phosphatase Cmp2 activated in response to ER stress (Mizuno et al, 2018). The Swd2/Nar1 interaction can be linked to the fact that Set1 754-1081 interacts with relatively good confidence with TyW1 according to hybrigenics criteria (see Methods), an iron-sulfur protein required for synthesis of wybutosine modified tRNA (Noma et al, 2006). In contrast to the stress genes mentioned above, many interactors involved in glucose metabolism (repression) interact only with the Set1 1-1081 or with members of the nSET module (Fig. S10).Reconstituted Set1C methylates the Snf2 RG motif within the AT-hook in vitroWe have shown above that SET1C interacts with Snf2C-AT-hook region (Figure 4). We thus tested whether the Snf2C-AT-hook could be methylated by Set1C. Set1C and truncated Set1C were reconstituted and affinity purified from Sf9 cells coinfected with baculoviruses that express FLAG-Set1 (or truncated Set1) and the seven other untagged subunits (Kim et al, 2013)(Fig. S3). We incubated reconstituted Set1C with the Snf2C, AT-hook and Bromo fragments in the presence of radioactive S-adenosylmethionine (3H-SAM). The results indicate that Set1C methylates in vitro purified Snf2C and Snf2C-AT-hook, but not Bromo (Fig. 8A, B) and that Snf2C-AT-hook methylation requires Set1 FL (Fig. 8C). Collectively these results show that the Snf2C-AT-hook (1384-1547) interacts and is methylated by Set1 FL in vitro. Lys 1494 (K1494) and Lys 1498 (K1498) located between the AT-hook domains of Snf2 were previously shown to be acetylated by Gcn5 (Kim et al, 2010). We individually mutated all the Lys of the Snf2-AT-hook into Arg and tested the methylation of the mutated fragments. null of the substitutions abolished or decreased the methylation of the AT-hook indicating that he AT-hook could be methylated on multiple sites or on other type of residues (Fig. S11). We thus sought to identify the minimal region within the Snf2-AT-hook that is methylated by Set1C in vitro. The Snf2-AT-hook domain was divided in 6 regions that were fused to the GST (Fig. 8D). In vitro methylation assays indicated that the Snf2-B1, B2, B3 were methylated by the reconstituted Set1C (Fig. 8E). As the Snf2-B3 fragment that contains the last four lysines (K1488, K1494, K1498, K1526) of Snf2-AT-hook domain was still methylated, it suggested that one of several of these lysines are methylated by Set1C. We then examined which of these 4 lysine residues are methylated by Set1C in vitro. To our surprise, mutating all the lysine residues of the Snf3-B3 fragment did not abolish the methylation of Snf3-B3 (Fig. 8F). These results led us to think that Set1C could methylate arginine residues, in particular those contained in the RGG motif of the Snf2-B3 fragment. We thus deleted the RG repeats of Snf2-B3 (Fig. 8G) and purified the Snf2-B3ΔRG. We found that deletion of the Snf2-B3 RG repeats abolished the interaction between Snf2-B3 and Set1 and methylation of Snf2-B3 (Fig. 8H, I). The fact that both, interaction and methylation are lost upon deletion of the RG motif argues in favor that Set1C, and not a potential contaminant from the reconstituted Set1C, is responsible for the methylation of the Snf2-B3 RG repeats. Individual deletion of pairs of arginine residues in the RG motif did not suppress methylation of the Snf2-B3 fragment, suggesting flexibility in Set1C’s ability to methylate Snf2-B3 (Fig. S12).Figure 8.Snf2 is methylated in tandem AT-hook domain by reconstituted Set1C.A) In vitro methyltransferase assay using purified SET1C and Snf2 fragments. H-SAM was used as a methyl-donor and methylated proteins were detected by autoradiography. The band marked with a red star is a degradation product of Snf2C. B) In vitro methyltransferase assay using SET1C and two Snf2-AT-hook fragments with two different tags. C) Schematic diagram of N-terminal truncated SET1 complexes (left) and in vitro methyltransferase assay with GST-Snf2-AT-hook and truncated SET1 complexes. D) Schematic diagram showing the positions of all lysines in Snf2-AT-hook and the further cleaved fragments of Snf2-AT-hook. The red box indicates the two lysines that are acetylated by Gcn5. E) Coomassie staining of purified Snf2 fragments (lower) and an in vitro methyltransferase assay using these fragments with SET1C (upper). F) In vitro methyltransferase assay by SET1C when each of the four lysines in the C-terminal region of the Snf2-AT-hook is substituted with arginine or when both lysines known to be acetylated by Gcn5 are substituted. G) A schematic diagram showing the position of the RG-repeat region and the design of Snf2-AT-hook with RG-repeat truncation. H and I) GST pull-down assay (H) and in vitro methyltransferase assay (I) using purified SET1C and GST-Snf2-AT-hook with or without RG-repeats.To further confirm that Snf2-B3 is methylated on arginine, we incubated a mutant Snf2-B3 (in which lysines were mutated in alanine to limit protease digestion prior mass spectroscopy analysis) with reconstituted Set1C and SAM and further purified the Snf2-B3 mutant on a Ni-NTA resin (Fig. 9A-D). We then analyzed by mass spectroscopy arginine methylation in Snf2-B3. We found that R1490, R1501, R1505, R1507 and R1517 were mono- and di-methylated while R1509, R1513, and R1519 were monomethylated (Fig. 9E). We thus confirmed that reconstituted Set1C has the ability to methylate in vitro multiple arginines in the Snf2B3 region.Figure 9.The arginines in the RG-repeat of Snf2 are methylated by reconstituted SET1C.A) A diagram showing the WT Snf2-B3 fragment and the Mut Snf2-B3 with all four lysines substituted with alanine. B) Coomassie staining of purified WT and Mut Snf2-B3. C) Mass-spectrometry experiment design to identify the methylation sites of Snf2-B3. D) Coomassie staining of Mut Snf2-B3 after methylation reaction and an additional purification step using Ni-NTA. The band corresponding to Mut Snf2-B3 (∼18 KDa) was excised and used for mass spectrometry analysis. The ∼37 KDa band observed in lanes 5–8 appears to be a SET1C subunit that binds non-specifically to Ni-NTA, likely SWD2 based on its size. E) Mass spectrometry analysis result of Snf2-B3 methylation sites revealed that multiple arginines in the RG-repeat were methylated. Amino acid sequence of Snf2 highlighting the arginines methylated by SET1. The sequence of the B3 fragment is shown in bold, and the arginines methylated by SET1 are marked in red. Methylation and demethylation are denoted as M and DM, respectively.Snf2 is methylated in vivo on its ARTSTRGR AT-hook motif in a Set1-dependent wayOur results show that an activity associated with reconstituted Set1C is capable of methylating arginines in the vicinity and in the RG repeats of Snf2C-AT-hook. The fact that only Set1FL is capable of methylating the AT-hook is in favor of this activity being associated with Set1C. Nevertheless, one cannot formally rule out that methylation the AT-hook is due to a contaminant from insect cells from which Set1C has been reconstituted.We thus investigated the interaction between Snf2 and Set1 in vivo and whether Snf2 is methylated by Set1C. We found that Set1 was coimmunoprecipitated with Snf2 tagged with a C-terminal Myc epitope (Fig. 10A). In contrast to what we observed in vitro, deletion of the RG motif of Snf2 did not affect the interaction between Snf2 and Set1, which is consistent with the fact that the SID of Snf2 is located upstream of the RG repeats (Fig. 10A).Figure 10.The arginines of the motif ARTSTRGR within the AT hook are methylated in vivo in a Set1-dependent way.A) Set1 interacts in vivo with Snf2 and Snf2-ΔRG. Myc-tagged Snf2 and Snf2ΔRG were immunoprecipitated with 9E10 Myc antibodies (see Methods) and revealed with either 9E10 (Upper panel) or Set1 antibodies (lower panel). B) Snf2C complex was purified from WT and set1Δ strains, separated on a 4-12% Bis-Tris Gel and Silver Stained (Left); the presence of Snf2-GFP is detected by Western-blotting with anti-GFP antibody (Right). The area corresponding to Snf2-GFP was excised from the gel and used for mass spectrometry analysis. Peptides flanking the RG repeats (1485-1549) with their PTM are shown in Fig. S14. C) Panel C show a focus on the amino sequence flanking the RG repeats. The positions of residues from D1485 to V1549 are indicated on the figure. Peptides identified after digestion of Snf2-GFP are indicated in color with their identified PTM indicated by the color code shown in the top of the panel. The small numbers above the amino acids indicate the probability of localisation according to the MS2 peaks. It should be noted that for wild-type K1488 and R1490, a peptide with dimethylation is detected, but no discriminating MS2 peak allows us to conclude whether K or R are dimethylated. This is also the case for dimethylation on R1528 and R1535. The results presented represent the observed PTMs from two independent experiments, each containing 3 replicates (Table S6).To further investigate Snf2 methylation by Set1C in vivo, we purified Snf2-GFP and its subunits with GFP nanobodies in the presence or absence of Set1 (Fig. 10B). The protein composition of the complex was determined by mass spectrometry in the presence of absence of Set1 (Figure 10B, Table S5). We recovered in both cases all the known components of the Snf2 complex (Snf2, Swi1, Swi3, Snf5, Snf12, Swp82, Arp9, Arp7, Snf6, Rtt102, Snf11, Taf14) (Table S5, Fig. S13A). Interestingly, we found that the arginine methyltransferase Rmt2 was found specifically enriched with the Snf2-GFP complex in set1Δ cells (Fig. S13).We then excised the Snf2-GFP gel band and subjected it to mass spectrometry. We examine the post-translational modifications (PTM) in the Snf2 (1485-1549) region in either WT or set1Δ strains (Table S6). We were unable to purify a peptide containing the RG repeats but we could identify the peptides flanking the RG motif (Fig. 10C). Fig. 10C represents the combined results of two independent experiments. In WT cells, we found that the region between K1494 and R1505 was reproducibly subjected to multiple PTM (Fig. 10C, upper panel). K1494 and K1498 that were previously found acetylated by Gcn5 (Kim et al, 2010) were found acetylated, however both lysines were also found to be methylated (Fig. 10 C). Of note H3K4 that is methylated by Set1C has been reported to be also acetylated by Gcn5 (Guillemette et al, 2011). T1995, S1996, S1999, T1502, S1503, and T1504 were found phosphorylated. Interestingly, R1501 was found mono-methylated while R1505 and R1507 were found di-methylated in the two independent Snf2-GFP purifications. These 3 arginines belong to a ARTSTRGR motif that lies in the Snf2-B3 fragment. They were also found to be methylated in vitro by the reconstituted Set1C (Fig. 9E). We then examine the posttranslational modifications of the peptides flanking the RG motif in the set1Δ strain (Fig. 10C, lower panel). Phosphorylation of serines and threonines was not modified in the set1Δ strain except for T1502. In the set1Δ strain, K1494 and K1498 remained methylated indicating that methylation of both lysines does not depend on Set1C. Strikingly, deleting SET1 abolished the mono-methylation of R1501 and the di-methylation of R1505, R1507 (Fig. 10C, lower panel). The results thus show that SET1C is required for the methylation of the three arginines of the ARTSTRGR motif, in agreement with the in vitro results.DiscussionWe have exploited the power of Y2H screening technology to establish an extended interaction network for the histone H3K4 methylase complex SET1C. Comprehensive data integration unveiled many potential functions of either the whole complex or individual subunits. This study is partly validated by the fact that we found interactors in known functions of SET1C, or putative functions for which the mechanism is unknown. We have also provided a number of validations by confirming the interaction through biochemical approaches. It seems likely that some proteins interact transiently with Set1 and its subunits, or under specific conditions of stress or nutrient limitations, explaining why these interactors have not previously been found associated with SET1C in biochemical approaches. The most illustrative case for this is the high-affinity interaction between Spp1 and Mer2 that takes place during the first meiotic division, which we had identified from the Spp1 screen and which has been extensively characterized (Acquaviva et al, 2013b). Nevertheless, considering the multitude of interactions identified, it seems unlikely that they all represent direct contacts, especially where multiple subunits of complexes or multiple components of a given metabolic pathways interact with Set1C. One caveat is that Y2H interactions can be mediated by endogenous proteins that bridge the interactions. But this does not make them less functionally significant. We anticipate that this systematic screen will be an invaluable resource for further investigation of the role of SET1C in known and novel processes revealed in this study. Processes discussed here include Set1’s export/import mechanism through the nucleus, interaction with RGG proteins some of which are targeted by arginine methyltransferase, cooperation of SET1C with chromatin remodeling factors in particular Snf2C, interaction with splicing factors notably with Prp8 and Prp22, coupling replication with histone deposition (Mcm2/Spp1), SET1C plasticity regulated by SUMOylation, and Ty element transposition. A recurrent question related to the identification of potential SET1C interactors is whether they could be methylated by SET1C. Based on the identification of domains that closely resembles the histone H3 modification site, we have shown that the Swd1 interactor Nrm1 is methylated by SET1C in vitro suggesting that proteins with H3K4-like domains represent a class of non-histone substrates that may be regulated by SET1C.We show that Set1C interacts both in vitro and in vivo with Snf2. Strikingly, reconstituted Set1C from insect cells is able to methylate in vitro the three arginines located within the ARTSTRGR motif of the AT-hook of Snf2. The in vivo results confirm that Set1 is required for the methylation of these arginines within the ARTSTRGR motif. It is interesting to note that this motif resembles the ARTKQTAR motif of the N-terminal H3. In yeast, H3R2 is mono- and di-methylated and its asymmetric di-methylation prevents H3K4me3 (Kirmizis et al, 2009, 2007) . In contrast symmetric di-methylation of H3R2 correlates with H3K4me3 and requires Set1 in vivo but not the classical arginine methyltransferases: Hmt1, Hmt2, Rkm2, Rkm3, Rkm4, Hsl7, and Efm1 (Li et al, 2015a; Yuan et al, 2012). It has been proposed that Set1C could be responsible for both H3R2me2s and H3K4me3 or that H3R2me2s deposited by a yet unidentified arginine methyltransferase requires the prior deposition of H3K4me3 (Yuan et al, 2012).Interestingly, previous tandem affinity protein purification of the mono- and asymmetric arginine methyltransferase Hmt1 (also termed Rmt1) identified 108 proteins associated with it (Jackson et al, 2012), a number of which were shown to be methylated. Snf2 was found to be copurified with Hmt1 in vivo and methylated in vitro by Hmt1 (Jackson et al, 2012) making Hmt1 a potential candidate for methylating Snf2. In vitro, purified Hmt1 catalyzes mono-methylation and asymmetric di-methylation of H3R2, however, deleting HMT1 does not affect the asymmetric di-methylation of H3R2 in vivo, which suggests that several methyltransferases could act redundantly on H3R2 (Li et al, 2015a). In mammals, H3R2 and H3R8 are asymmetrically di-methylated by PRMT6 (Hamey et al, 2021; Guccione et al, 2007; Hyllus et al, 2007). It has been also reported that PRMT5 can be found associated with hSWI/SNF and has the ability to methylate H3R8 (Pal et al, 2004). We did not identify Rmt1 in our purification but found that the arginine mono-methyl transferase Rmt2 enriched with Snf2-GFP in set1Δ cells. Rmt2 is a type IV methyl transferase that was reported to methylate the ribosomal protein L12 (Low & Wilkins, 2012; Chern et al, 2002). We could envision the possibility the possibility that Set1C could cooperate with distinct protein arginine methyl transferases to promote the mono-methylation of R1501 and the di-methylation R1505 and R1507 within the ARTSTRGR motif. However, it was previously reported that the combined inactivation of Rmt1, Rmt2 and Hsl7 did not affect the mono-methylation of H3R2, weakening this hypothesis (Kirmizis et al, 2009). It is possible that in yeast there is a redundancy of enzymes capable of methylating the Snf2 ARTSTRGR motif, but that for each of them their activity on this motif depends on the presence of Set1. Alternatively, Set1C could directly methylate the ARTSTRGR motif as discussed for the ARTKQTAR H3 motif (Yuan et al, 2012). The fact that Set1C interacts with Snf2, Gbp2, Nop1, Nab2, Dbb1 (Fig. 2), all of which have RG motifs and are mostly Hmt1 substrates, raises the possibility of a general interplay between methylarginine and methyllysine.Interestingly, a very recent article shows that PRMT1 binds to the N-terminal region of MLL2 and methylates multiple arginine residues within its RGG/RG motifs (An et al, 2025).Materials and methodsStrain constructionAll strains and plasmids used in this study are listed in Table S1. To obtain gene deletions and expression of tagged proteins, we amplified by PCR a disruption or a tagging cassette containing the appropriate marker as described (Janke et al, 2004).Yeast two-hybrid analysisYeast two-hybrid screening was performed by Hybrigenics Services, S.A.S., Evry, France (73). The coding sequence for Set1-FL, Set1 1-754, Set1 754-1081 and Bre2 were cloned into pB66 (GAL4-bait) as C-terminal fusions to the Gal4 DNA-binding domain while those of Swd1, Swd3, Sdc1, Spp1, and Shg1 were cloned into pB27 as a C-terminal fusion to LexA (LEXA-bait). SWD2 was cloned into pB43 as a N-terminal fusion (SWD2-GAL4). Individual bait cloning was performed by using specific primers for PCR for every bait. Every PCR fragment subcloned as bait is entirely sequenced to avoid eventual mismatches in the coding sequence. The constructs were used as baits to screen a genomic S. cerevisiae library constructed into pP6 based on the pGADGH plasmid (Wilson et al, 1993). pB6, pB66 and pB43 are derived from the original pAS2Δ vector (Fromont-Racine et al, 1997). The library was submitted to very strict quality controls. Each protein is represented by several fragments (domains) and the library has been screened with a set of 6 reference baits before any other bait proteins are screened.The Gal4 constructs were screened using a mating approach with YHGX13 (Y187 ade2-101::loxP-kanMX-loxP, MATalpha) and CG1945 (MATa) yeast strains. The LexA constructs were screened using a mating approach with YHGX13 (Y187 ade2-101::loxP-kanMX-loxP, mat alpha) and L40ΔGal4 (MATa) yeast strains as previously described (Fromont-Racine et al, 1997). Positive clones were selected on a medium lacking tryptophan, leucine and histidine and supplemented with 3-aminotriazole (3AT) if necessary to handle bait auto-activation. The prey fragments of the positive clones were amplified by PCR and sequenced at their 5’ and 3’ junctions. The resulting sequences were used to identify the corresponding interacting proteins in the GenBank database (NCBI) using a fully automated procedure.A confidence score (PBS, for Predicted Biological Score) was attributed to each interaction as previously described (Formstecher et al, 2005). The confidence score relies on two different levels of analysis. Firstly, a local score takes into account the redundancy and independency of prey fragments, as well as the distribution of reading frames and stop codons in overlapping fragments. Secondly, a global score takes into account the interactions found in all the screens performed at Hybrigenics using the same library. This global score represents the probability of an interaction being nonspecific. For practical use, the scores were divided into four categories, from A (highest confidence) to D (lowest confidence). A fifth category (E) specifically flags interactions involving highly connected prey domains previously found several times in screens performed on libraries derived from the same organism. Finally, several of these highly connected domains have been confirmed as false-positives of the technique. The PBS scores have been shown to positively correlate with the biological significance of interactions. When possible, the bait interacting domain of each prey is provided. The PBS scores have been shown to positively correlate with the biological significance of interactions (Rain et al, 2001). e-values for the interactions are available in the Hybrigenics database.Protein expression and protein interaction assaysProtein expression was done in Escherichia coli BL21 cells and purification of GST-fusion proteins were as described (Dichtl et al, 2002). MBP-fusion proteins were purified according to manufacturer’s instructions (New England BioLabs, Beverly, MA, USA). For His-tagged proteins expressed in bacteria, cDNAs were inserted into the pET28 vector (Novagen), expressed in Escherichia coli, and purified using Ni-NTA beads (Qiagen) following the previously described procedure (Kim & Roeder, 2011). GST pull-down assays were done as described (Dichtl et al, 2002).A baculovirus expression system was used to express and reconstitute Set1 complexes containing either FLAG-Set1 or FLAG-Set1-C762. cDNAs were subcloned into pFASTBAC1, with or without an epitope tag, and baculoviruses were produced following the manufacturer’s instructions (Gibco-Invitrogen). Sf9 cells were infected with various combinations of baculoviruses, and the complexes were purified by affinity chromatography using M2 agarose, as previously described (Kim & Roeder, 2011).TAP-Nis1 affinity purification and mass spectrometry analysisTAP-Nis1 and control cells (W303a) were grown in YPD, harvested in logarithmic phase (OD600 0.6-0.85) and cryo-lysed as previously described (Trahan & Oeffinger, 2022). Affinity purification was performed in RNP100 (20 mM HEPES-KOH pH 7.4, 100 mM NH4OAc, 0.5% Triton X-100, 0.1% Tween 20, 1:100 solution P, 1:5000 antifoam A, 100 mM NaCl) as described in (Trahan & Oeffinger, 2022). Following washes, the samples were on-bead trypsin digested in a volume of 50µl (f. c. 20 μg/mL trypsin (Sigma, proteomics grade) in 20 mM Tris-HCl (pH 8.0), 37 °C, 900 rpm, 16-20 h; stopped with 2% formic acid) and analyzed by tandem mass spectrometry as described in (Trahan & Oeffinger, 2022) using a 70-minute gradient on a LTQ Orbitrap Velos is a hybrid mass spectrometer (ThermoFisher Scientific) in Data-dependent mode. Data were processed with Thermo Excalibur to generate a raw file. Mascot search server (53 Version 2.3.02) was used with a parent tolerance of 10 ppm for precursor ions, 0.52Da for fragments, and only considering 1 possible missed cleavage as well as a mass change of +16 for methionine oxidations in the mass calculation. Data were searched against S. cerevisiae NCBI database and analyzed in Scaffold (version 3.6.4). The threshold and false discovery rates (FDR) were set to 80% and 0.37% respectively for peptides, and to 95% (1 peptide minimum) and 1.9% for respectively for proteins. Exclusive spectrum counts (ESC) were used for analysis and for each prey, the highest values obtained in the controls were removed from those of the samples during analysis.In vitro methylationIn vitro methylation reactions were done essentially as described (Roguev et al, 2001). 30 ml reactions typically contained 2 to 4 ml of partially purified SET1C complex, 2 to 4 mg substrate (either peptide, or core histone mixture, or recombinant H3) and 2 ml S-adenosyl (methyl-3H) methionine in MTA buffer (50 mM Tris 8.5, 20 mM KCl, 10 mM MgCl2, 250 mM sucrose). Reactions with core histones and recombinant H3 were resolved on 4-20% NuPAGE gels and subjected to fluorography. For Snf2 methylation assays, reaction mixtures containing purified SET1C (with 30 ng of the Bre2 subunit) and 200 ng of Snf2 fragments in 20 μl of reaction buffer (25 mM HEPES [pH 7.6], 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 10% glycerol) were supplemented with 1 μCi of S-adenosyl (methyl-3H) methionine (PerkinElmer) and incubated at 30°C for 1 hour. Proteins were resolved by SDS-PAGE and subjected to fluorography. For fluorography, gels were fixed for 30 min in 40% methanol, 10% acetic acid, treated with EN3HANCE solution (Perkin Elmer) for 60 min, washed in cold dH20 for 30 min, dried and exposed for five to fourteen days to photographic film.Mass spectrometry-based identification of Arg methylation in Snf2-B3Mutant Snf2-B3 underwent a methylation reaction with SET1C and SAM, followed by purification via Ni-NTA affinity chromatography and separation by SDS-PAGE. The gel bands corresponding to the mutant Snf2-B3 were excised and subjected to in-gel digestion with AspN enzyme, followed by peptide extraction. The resulting peptide fractions were analyzed using an Easy-nLC 1200 coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, MA, USA) at the Korea Basic Science Institute (Ochang). Peptides were separated on a C18 column using a 150-minute gradient, and data were acquired in data-dependent acquisition (DDA) mode. Full MS scans were acquired in the Orbitrap at a resolution of 60,000 over an m/z range of 350–2,000. Precursors for MS/MS analysis were selected for higher-energy collisional dissociation (HCD) fragmentation at a normalized collision energy of 30%. Raw MS data were processed using Proteome Discoverer with the SEQUEST search engine. The data were searched against the sequence of Snf2-B3 with a precursor mass tolerance of 10 ppm and a fragment mass tolerance of 0.02 Da. AspN was set as the digestion enzyme, allowing for up to two missed cleavages. Methylation, dimethylation, and methionine oxidation were specified as variable modifications. The false discovery rate (FDR) was set to 1% at the peptide level.Y2H interaction of Set1 fragments with selected preysThe Set1 fragments were amplified from the pB66-Set1-FL plasmid and cloned into the SfiI site of pB66. The selected preys (Snf2, Prp8, and Prp22) into p6 (Hybrigenics) were extracted from the screen. Plasmid pB66 contains the Gal4 DNA binding domain and the TRP1 marker while pP6 expresses the Gal4 activating domain and the LEU2 marker. TOTO cells were transformed with the different pB66-Set1-Fragments and the pP6-interactors and incubated 3 days at 30°C on SD-LEU-TRP. The transformant colonies were then streaked on SD-LEU-TRP-HIS containing either 5 mM or 20 mM of 3AT. To visualize interaction between Set1 fragments and the interactors, yeast cells were incubated 2 days at 30°C and cell growth was examined.CO-IP of Prp22-FLAG with Myc-Set1Co-immunoprecipitation experiment was performed in W303 expressing chromosomally encoded Myc-Set1 (Dehe et al, 2006) and Prp22AID-FLAG (Mendoza-Ochoa et al, 2019). 250 ml of culture at an OD600 of 0.8 was harvested by centrifugation at 1000 x g and washed twice in ice-cold 1 X PBS. The cell pellet was re-suspended in 900 µl lysis buffer (50 mM Tris-HCl pH 7.5, 2 mM Mg2Cl2, 150 mM NaCl, 0.2% NP-40 and one complete EDTA-free proteinase inhibitor tablet (Roche #11836145001) and 400 µl zirconia beads. Cells were lysed using a Mini-Beadbeater-24 (BioSpec Products) twice at 2000 rpm for 2 min followed by 2 min on ice. The sample was centrifuged at 1000 x g for 2 min, the supernatant was collected and additionally centrifuged at 20,000 x g for 30 min at 4℃ and used for immunoprecipitation. The concentration of protein was measured using the Bradford assay, and 1 mg of protein used per IP. Prior to immunoprecipitation, extract was pre-cleared by adding ½ volume unconjugated Protein A/G Dynabeads (Life Technologies ##10001D/10003D). 50 µl Protein A/G Dynabeads conjugated to antibody were incubated with the appropriate volume of extract on a rotating wheel overnight at 4℃. The next day, beads were washed 8 times in lysis buffer (non-bound fraction kept for analysis). 20 µl of loading buffer was added to the beads, input and non-bound samples, which were boiled for 10 min before loading on a NuPAGE 4-12% Bis-Tris gel Bis-Tris (Invitrogen) and western blotting was performed.Set1 SUMOylation analysisCells were transformed with a plasmid encoding 6His-SUMO under the CUP1 promoter (YEp352-6His-SUMO) or the corresponding empty vector (Niño et al, 2016). The transformed cells were grown on selective medium and stimulated overnight with 0.1 mM CuSO4. 200 OD600 of cells were collected and lysed with glass beads in a 20% TCA solution, the final TCA concentration was adjusted to 12%. Cell lysates were incubated at 4°C during 45 min, and precipitated proteins were collected by centrifugation. Proteins were resuspended in loading buffer (6 M guanidinium-HCl, 100 mM KH2PO4, 20 mM Tris-HCl, pH 8,0, 100 mM NaCl, 0,1% Triton X-100, and 10 mM imidazole). 6His-SUMOylated proteins were purified using Ni-NTA agarose beads (Qiagen), pre-equilibrated with loading buffer, and incubated for 1 h at room temperature. After incubation the beads were collected by centrifugation (3000 rpm, 2min) and washed twice with wash buffer (8 M urea, 100 mM Na2HPO4/NaH2PO4 pH 6,4, 10 mM Tris–HCl, pH 6.4, 10 mM Imidazole, 10 mM β-mercaptoethanol, and 0.1% Triton X-100). 6His-SUMOylated proteins were eluted with 50 ml 2X laemmli buffer (95°C, 5min). The proteins in the eluted fraction were analyzed by Western blot using anti-MYC (for endogenous Myc-Set1), anti-GAL4 (for GBD-Set1-full length and fragments) and polyclonal rabbit anti-Smt3.Interaction Snf2-AT-hook with SET1C and SET1C-762GST-tagged Snf2 fragments (final concentration 250 nM) were combined with either the full-length SET1C or C762SET1C (final concentration 25 nM), along with 15 µl of glutathione-Sepharose 4B resin and BSA (final concentration 0.2 mg/ml) in a binding buffer containing 20 mM Tris-Cl (pH 7.9), 150 mM NaCl, 0.2 mM EDTA, 20% glycerol, 0.1% NP-40, and 1 mM PMSF, making a total volume of 300 µl. The mixtures were rotated at 4°C for 3 hours, followed by four washes with the binding buffer. The proteins bound to the resin were then eluted, separated by SDS-PAGE, and analyzed by immunoblotting.CO-IP of Snf2-Myc and Snf2ΔRG-Myc with Set1WT cells or expressing Snf2-Myc and Snf2ΔRG-Myc (200 ml) were grown at 30°C in YPD to and O.D600 = 0.8. Cells were collected by centrifugation, washed once with 10 mM Tris-HCL, pH 8.0 and snap-frozen in liquid nitrogen. Cells were lysed using Retsch with the following parameters :2 times 2 min at 30 m/S. The powder was resuspended in 3 ml of TMG 50 (10 mM Tris-HCL, pH 8.0, 0,1 mM MgCl2, 10% (V/V) glycerol, 50 mM NaCl, 0.1 mM EDTA, 0.1 mM DTT) containing protease inhibitors, 10 mM MG-132 and 1 mM PMSF. Cell lysates were clarified by centrifugation (13 300 rpm, 15’, 4°C) and 1.4 ml of supernatant were recovered. Protein concentration was determined using nanodrop and samples were adjusted to the same concentration by adding lysis buffer. Immunoprecipitation was performed overnight with 5 ml of 9E10 antibody (Santa Cruz Biotechnology) following by an incubation for 3 hours with 25 ml of pre-equilibrated protein-G dynabeads (Invitrogen). Immunoprecipitations were washed 3X with TMG 50 buffer and eluted using 25 ml of 1X Laemmli loading Buffer. Samples were resolved on a 7.5% Acrylamide gel, transferred on a nitrocellulose membrane and reveal with either anti-Myc 9E10 (Santa Cruz Biotechnology) and anti-Set1 antibodies.Snf2-GFP Complex purification800 ml of Snf2-GFP and Snf2-GFP set1Δ cells were grown at 30°C in YPD to O.D600 = 0.8. Cell pellets were treated as described above except that supernatants were incubated with 25 ml of GFP nanobody coated Dynabeads (Gift from M. Modesti, CRCM Marseille) for 250 min, wash 3X with TMG 50 buffer and eluted in 30 ml of 2X Laemmli Loading buffer containing 50 mM DTT. For Western blotting, 1 ml was resolved on a NuPAGE 4-12% Bis-Tris Gel (Invitrogen), transferred on a nitrocellulose membrane and revealed using an anti-GFP antibody (A-11122-Invitrogen). For the Silver Staining, 2,5 ml were resolved on a NuPAGE 4-12% Bis-Tris Gel (Invitrogen) and stained using PierceTM Silver Stain kit (Thermo Scientific).Mass spectrometry-based identification of post-translational modifications in Snf2-GFPPurified Snf2-GFP (n=3, biological replicates) from WT and set1Δ strains were loaded on NuPAGE™ 4–12% Bis–tris acrylamide gels according to the manufacturer’s instructions (Life Technologies). Running of protein was stopped as soon as proteins stacked in a single band. Protein containing bands were stained with Imperial Blue (Pierce), cut from the gel and digested with high sequencing grade trypsin (Promega) before mass spectrometry analysis. Briefly, gel pieces were washed and destained using few steps of 100 mM NH4HCO3. Destained gel pieces were shrunk with 100 mM ammonium bicarbonate in 50% acetonitrile and dried at RT. Protein spots were then rehydrated using 10mM DTT in 25 mM ammonium bicarbonate pH 8.0 for 45 min at 56°C. This solution was replaced by 55 mM iodoacetamide in 25 mM ammonium bicarbonate pH 8.0 and the gel pieces were incubated for 30 min at room temperature in the dark. They were then washed twice in 25 mM ammonium bicarbonate and finally shrunk by incubation for 5 min with 25 mM ammonium bicarbonate in 50% acetonitrile. The resulting alkylated gel pieces were dried at room temperature. The dried gel pieces were reswollen by incubation in 25 mM ammonium bicarbonate pH 8.0 supplemented with 12.5 ng/µl trypsin (Promega) for 1h at 4°C and then incubated overnight at 37°C. Peptides were harvested by collecting the initial digestion solution and carrying out two extractions; first in 5% formic acid and then in 5% formic acid in 60% acetonitrile. Pooled extracts were dried down in a centrifugal vacuum system. Samples were reconstituted in 0.1% TFA 4% acetonitrile before mass spectrometry using an Orbitrap Fusion Lumos Tribrid Mass Spectrometer (ThermoFisher Scientific, San Jose, CA) online with an Ultimate 3000RSLCnano chromatography system (ThermoFisher Scientific, Sunnyvale, CA). Peptides were separated at 40°C using a two steps linear gradient (4–20% acetonitrile/H2O; 0.1% formic acid for 110 min and 20-32% acetonitrile/H2O; 0.1% formic acid for 10 min). An EASY-Spray nanosource was used for peptide ionization (2,200 V, 275°C). MS was conducted using a data-dependent acquisition mode (DDA). The Orbitrap Lumos was used in data dependent mode to switch consistently between MS and MS/MS. Time between Masters Scans was set to 3 seconds. MS spectra were acquired with the Orbitrap in the range of m/z 400-1600 at a FWHM resolution of 120 000 measured at 400 m/z. AGC target was set at 4.0e5 with a 50 ms Maximum Injection Time. For internal mass calibration the 445.120025 ion was used as lock mass. The more abundant precursor ions were selected and collision-induced dissociation fragmentation was performed in the ion trap to have maximum sensitivity and yield a maximum amount of MS/MS data. Number of precursor ions was automatically defined along run in 3s windows using the “Inject Ions for All Available parallelizable time option” with a maximum injection time of 300 ms. The signal threshold for an MS/MS event was set to 5000 counts. Charge state screening was enabled to exclude precursors with 0 and 1 charge states. Dynamic exclusion was enabled with a repeat count of 1 and duration of 60 s.For identification of PTMs the region corresponding to Snf2-GFP was excised from the gel and subjected to enzymatic digestion under the same conditions as above. Raw mass spectrometry files were analysed using MaxQuant software (version 1.6.3.4) as above with the exception of the Max Missed cleavage set at 5, the FDR at peptide and proteins levels at 5% and with the following variable modifications activated; Lysine acetylation (+42.0106); Serine, threonine and tyrosine phosphorylation (+79.966); Lysine and arginine methylation (+14.0157), dimethylation (+28.0313) and trimethylation (+42.0470).Data Processing ProtocolRelative intensity-based label-free quantification (LFQ) was processed using the MaxLFQ algorithm from the freely available MaxQuant computational proteomics platform, version 1.6.2.1 (Cox et al. 2014, Cox et al. 2008). The acquired raw LC Orbitrap MS data were first processed using the integrated Andromeda search engine (Cox et 2011). Spectra were searched against the Saccharomyces cerevisiae (organism ID 4932) extracted from UniProt containing 7904 entries. The following parameters were used for searches: (i) trypsin allowing cleavage before proline; (ii) two missed cleavage was allowed; (iii) cysteine carbamidomethylation (+57.02146) as a fixed modification and methionine oxidation (+15.99491) and N-terminal acetylation (+42.0106) as variable modifications. The match between runs option was enabled. The false discovery rate (FDR) at the peptide and protein levels were set to 1% and determined by searching a reverse database. For protein grouping, all proteins that cannot be distinguished based on their identified peptides were assembled into a single entry according to the MaxQuant rules. The statistical analysis was done with Perseus program (version 1.6.1.3) (Tyanova and Cox 2018) from the MaxQuant environment (www.maxquant.org). Quantifiable proteins were defined as those detected in above 70% of samples in one condition or more. Protein LFQ normalized intensities were base 2 logarithmized to obtain a normal distribution. Missing values were replaced using data imputation by randomly selecting from a normal distribution centered on the lower edge of the intensity values that simulates signals of low abundant proteins using default parameters (a downshift of 1.8 standard deviation and a width of 0.3 of the original distribution). The protein composition of the complex was determined using, a two-sample t-test using permutation-based FDR-controlled at 0.01 and employing 250 permutations and a scaling factor s0 with a value of 3. All proteins passing these criteria with a positive fold change are considered to be the potential interactome of Snf2.AntibodiesFor IP: Mouse anti-FLAG (Sigma M2 #F1804); 10 μl/IP with Dynabeads and Protein G. For westerns - primary antibodies: Mouse anti-c-MYC (Santa Cruz #SC-40x) 1:1000; Rat anti-FLAG (Agilent #200474) 1:1000; Mouse anti-Gal4 Binding Domain (Euromedex) 1:5000; Rabbit anti-Smt3 is a gift from B. Palancade (Institut Jacques Monod, Paris). Anti-Bre2 is a gift from Peter Nagy. The rabbit anti-Spp1 antibody was developed in V. Géli’s lab. The anti-Spp1 antibody is showing significant cross-reactivity with other cellular proteins but did allow the identification of Spp1 in purified and partially purified fractions. Secondary antibodies: Goat anti-mouse IRDye680RD (LI-COR #926-68070) 1:10,000, Goat anti-rat IRDye800RD (LI-COR #926-32219) 1:10,000.Data availabilityAdditional data and reagents are available upon request to the corresponding authors. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (www.proteomexchange.org) via the PRIDE partner repository (https://www.ebi.ac.uk/pride/login): Snf2-B3 data (accession number PXD061448), Snf2-GFP complex (accession number PXD061496), Snf2 PTM (accession number PXD061531).AcknowledgementsWe are grateful to Sandra Holbein and Andre Halbach for help with MBP-Nrm1 fusion proteins. We thank Benoit Palancade for his help in the interpretation of the Y2H results and reagents.Additional informationFundingThis work is supported by the “Ligue Nationale Contre le Cancer” (LNCC) (Equipe labellisée, GELI/2019). Funding to B.D. was provided by grants from the Swiss National Science Foundation (PP00A--102941/1 and 31003A_1327010). I.E.M. was funded by Wellcome Trust PhD Studentship (105256) and work in the Wellcome Centre for Cell Biology was supported by Wellcome core funding (092076). Funding to M. O. was provided by the Natural Sciences and Engineering Research Council of Canada to M.O. (RGPIN-2020-06924). Funding to J. K was provided by the National Research Foundation of Korea (NRF-2022R1A2C3012803). Y. H. K is supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. RS-2024-00454407). Proteomics analyses using the mass spectrometry facility of Marseille Proteomics (marseille-proteomique.univ-amu.fr) are supported by IBISA, the Cancéropôle PACA, the Provence-Alpes-Côte d’Azur Region, the Institut Paoli-Calmettes, and Fonds Européens de Développement Regional (FEDER).Author contributionsB.D. J. K and V.G. designed the study and secured the funding. All the Y2H screens and prey identification were performed by Hybrigenics in close interaction with B.D. and V. G. P.L. performed molecular cloning, contributed to validation experiments, and purified Snf2-GFP for subsequent analyses. SET1C and SET1C-C762 purification and interaction assays with Snf2-AT-hook were performed by J. K. and K.P. Set1 SUMOylation experiments were performed by C. N. M. D and B. D. organized the results shown in Table S2. L. L constructed the F1-F5 fragments and contributed to validation experiments. Pull-down experiments and Nrm1 methylation assay were performed by B. D. TAP-Nis1 data were provided by M.O. I.M. and J. B. performed and discuss the Set1/Prp22 interaction. D.K. P and H. H. K performed mass spectrometry analysis of the Snf2-B3. S.A. and L.C. performed mass spectrometry analysis of the Snf2-GFP. V.G. and B. D. wrote the manuscript with inputs of the authors.FundingLigue Contre le Cancer (laliguecancer) (GELI/2019)Vincent GeliSwiss Re | Swiss Re Foundation (SRF) (PP00A--102941/1)Bernhard DichtlWellcome Trust (WT)https://doi.org/10.35802/105256Isabella E MaudlinNatural Sciences and Engineering Research Council of Canada (NSERC) (RGPIN-2020-06924)Marlene OeffingerNational Research Foundation of Korea (NRF) (NRF-2022R1A2C3012803)Jaehoon KimNational Research Foundation of Korea (NRF) (RS-2024-00454407)Young Hye KimAdditional filesSupplementary information.Table S2. All SET1C interactors identified in the 10 Y2H screens. The interactors common to several subunit and to common Set1 fragments are shown on sheets 12 and 13 of the table, respectively.Table S3. Mass spectrometry analysis of TAP-Nis1 affinity purification.Table S4. H3K4-like domain proteins. We used the scansite search algorithm (http://scansite.mit.edu) and systematically identified sequence motifs that are related to the Set1C modification site in histone H3.Table S5. Snf2-GFP complex in WT and set1∆ cells.Table S6. Snf2 post-translational modifications in either WT or set1∆ strains.ReferencesAcquaviva LDrogat JDehé P-Mde La Roche Saint-André CGéli V2013aSpp1 at the crossroads of H3K4me3 regulation and meiotic recombinationEpigenetics 8:355–360Google ScholarAcquaviva LSzekvolgyi LDichtl BDichtl BSSaint Andre C d LRNicolas AGeli V2013bThe COMPASS Subunit Spp1 Links Histone Methylation to Initiation of Meiotic RecombinationScience 339:215–218Google ScholarAdam CGuérois RCitarella AVerardi LAdolphe FBéneut CSommermeyer VRamus CGovin JCouté Yet al2018The PHD finger protein Spp1 has distinct functions in the Set1 and the meiotic DSB formation complexesPLoS Genet 14:e1007223Google ScholarAn DKim JMoon BKim HNguyen HPark SLee JEKim J-AKim J2025PRMT1-mediated methylation regulates MLL2 stability and gene expressionNucleic Acids Res 53Google ScholarAn SYoon JKim HSong J-JCho U-S2017Structure-based nuclear import mechanism of histones H3 and H4 mediated by Kap123eLife 6:e30244https://doi.org/10.7554/eLife.30244Google ScholarArigo JTCarroll KLAmes JMCorden JL2006Regulation of yeast NRD1 expression by premature transcription terminationMol Cell 21:641–651Google ScholarBae HJDubarry MJeon JSoares LMDargemont CKim JGeli VBuratowski S2020The Set1 N-terminal domain and Swd2 interact with RNA polymerase II CTD to recruit COMPASSNat Commun 11:2181Google ScholarBaker RPHarreman MTEccleston JFCorbett AHStewart M2001Interaction between Ran and Mog1 is required for efficient nuclear protein importJ Biol Chem 276:41255–41262Google ScholarBalk JPierik AJNetz DJAMühlenhoff ULill R2004The hydrogenase-like Nar1p is essential for maturation of cytosolic and nuclear iron-sulphur proteinsEMBO J 23:2105–2115Google ScholarBattaglia SLidschreiber MBaejen CTorkler PVos SMCramer P2017RNA-dependent chromatin association of transcription elongation factors and Pol II CTD kinaseseLife 6:e25637https://doi.org/10.7554/eLife.25637Google ScholarBeck MHurt E2017The nuclear pore complex: understanding its function through structural insightNat Rev Mol Cell Biol 18:73–89Google ScholarBeilharz THHarrison PFMiles DMSee MMLe UMMKalanon MCurtis MJHasan QSaksouk JMargaritis Tet al2017Coordination of Cell Cycle Progression and Mitotic Spindle Assembly Involves Histone H3 Lysine 4 Methylation by Set1/COMPASSGenetics 205:185–199Google ScholarBerretta JPinskaya MMorillon A2008A cryptic unstable transcript mediates transcriptional trans-silencing of the Ty1 retrotransposon in S. cerevisiaeGenes Amp Dev 22:615–626Google ScholarBorde VRobine NLin WBonfils SGéli VNicolas A2008Histone H3 lysine 4 trimethylation marks meiotic recombination initiation sitesEMBO J 28:99–111Google ScholarBriggs SDBryk MStrahl BDCheung WLDavie JKDent SYWinston FAllis CD2001Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiaeGenes Amp Dev 15:3286–3295Google Scholarde Bruin RAMKalashnikova TIChahwan CMcDonald WHWohlschlegel JYates JRussell PWittenberg C2006Constraining G1-specific transcription to late G1 phase: the MBF-associated corepressor Nrm1 acts via negative feedbackMol Cell 23:483–496Google ScholarBryk MBriggs SDStrahl BDCurcio MJAllis CDWinston F2002Evidence that Set1, a factor required for methylation of histone H3, regulates rDNA silencing in S. cerevisiae by a Sir2-independent mechanismCurr Biol CB 12:165–170Google ScholarCastelnuovo MRahman SGuffanti EInfantino VStutz FZenklusen D2013Bimodal expression of PHO84 is modulated by early termination of antisense transcriptionNat Struct Mol Biol 20:851–858Google ScholarCastelnuovo MZaugg JBGuffanti EMaffioletti ACamblong JXu ZClauder-Münster SSteinmetz LMLuscombe NMStutz F2014Role of histone modifications and early termination in pervasive transcription and antisense-mediated gene silencing in yeastNucleic Acids Res 42:4348–4362Google ScholarCenik BKShilatifard A2021COMPASS and SWI/SNF complexes in development and diseaseNat Rev Genet 22:38–58Google ScholarCheng HHe XMoore C2004The Essential WD Repeat Protein Swd2 Has Dual Functions in RNA Polymerase II Transcription Termination and Lysine 4 Methylation of Histone H3Mol Cell Biol 24:2932–2943Google ScholarChern M-KChang K-NLiu L-FTam T-CSLiu Y-CLiang Y-LTam MF2002Yeast Ribosomal Protein L12 Is a Substrate of Protein-arginine Methyltransferase 2J Biol Chem 277:15345–15353Google ScholarChoi JRajagopal AXu Y-FRabinowitz JDO’Shea EK2017A systematic genetic screen for genes involved in sensing inorganic phosphate availability in Saccharomyces cerevisiaePloS One 12:e0176085Google ScholarChong SYCutler SLin J-JTsai C-HTsai H-KBiggins STsukiyama TLo Y-CKao C-F2020H3K4 methylation at active genes mitigates transcription-replication conflicts during replication stressNat Commun 11:809Google ScholarChoudhury RSingh SArumugam SRoguev AStewart AF2019The Set1 complex is dimeric and acts with Jhd2 demethylation to convey symmetrical H3K4 trimethylationGenes Dev 33:550–564Google ScholarCorda YSchramke VLonghese MPSmokvina TPaciotti VBrevet VGilson EGeli V1999Interaction between Set1p and checkpoint protein Mec3p in DNA repair and telomere functionsNat Genet 21:204–208Google ScholarCôté JQuinn JWorkman JLPeterson CL1994Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complexScience 265:53–60Google ScholarDaicho KKoike NOtt RGDaum GUshimaru T2020TORC1 ensures membrane trafficking of Tat2 tryptophan permease via a novel transcriptional activator Vhr2 in budding yeastCell Signal 68:109542Google ScholarDehe PMDichtl BSchaft DRoguev APamblanco MLebrun RRodriguez-Gil AMkandawire MLandsberg KShevchenko Aet al2006Protein Interactions within the Set1 Complex and Their Roles in the Regulation of Histone 3 Lysine 4 MethylationJ Biol Chem 281:35404–35412Google ScholarDehé P-MGéli V2006The multiple faces of Set1Biochem Cell Biol Biochim Biol Cell 84:536–548Google ScholarDelamarre ABarthe Ade la Roche Saint-André CLuciano PForey RPadioleau ISkrzypczak MGinalski KGéli VPasero Pet al2020MRX Increases Chromatin Accessibility at Stalled Replication Forks to Promote Nascent DNA Resection and Cohesin LoadingMol Cell 77:395–410Google ScholarDeshpande NBryk M2023Diverse and dynamic forms of gene regulation by the S. cerevisiae histone methyltransferase Set1Curr Genet 69:91–114Google ScholarDeshpande NJordan RHenderson Pozzi MBryk M2022Histone 3 lysine 4 monomethylation supports activation of transcription in S. cerevisiae during nutrient stressCurr Genet 68:181–194Google ScholarDichtl B2004Functions for S. cerevisiae Swd2p in 3’ end formation of specific mRNAs and snoRNAs and global histone 3 lysine 4 methylationRNA 10:965–977Google ScholarDichtl BBlank DSadowski MHübner WWeiser SKeller W2002Yhh1p/Cft1p directly links poly(A) site recognition and RNA polymerase II transcription terminationEMBO J 21:4125–4135Google Scholarvan Dijk ELChen CLd’Aubenton-Carafa YGourvennec SKwapisz MRoche VBertrand CSilvain MLegoix-Né PLoeillet Set al2011XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeastNature 475:114–117Google ScholarDover JSchneider JTawiah-Boateng MAWood ADean KJohnston MShilatifard A2002Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6J Biol Chem 277:28368–28371Google ScholarD’Urso ATakahashi Y-HXiong BMarone JCoukos RRandise-Hinchliff CWang J-PShilatifard ABrickner JH2016Set1/COMPASS and Mediator are repurposed to promote epigenetic transcriptional memoryeLife 5:111Google ScholarFaucher DWellinger RJ2010Methylated H3K4, a Transcription-Associated Histone Modification, Is Involved in the DNA Damage Response PathwayPLoS Genet 6:e1001082Google ScholarFormstecher EAresta SCollura VHamburger AMeil ATrehin AReverdy CBetin VMaire SBrun Cet al2005Protein interaction mapping: a Drosophila case studyGenome Res 15:376–384Google ScholarFromont-Racine MRain JCLegrain P1997Toward a functional analysis of the yeast genome through exhaustive two-hybrid screensNat Genet 16:277–282Google ScholarGhaddar NCorda YLuciano PGalli MDoksani YGéli V2023The COMPASS subunit Spp1 protects nascent DNA at the Tus/Ter replication fork barrier by limiting DNA availability to nucleasesNat Commun 14:5430Google ScholarGiaever GLissina ENislow C2019Network dynamics of the yeast methyltransferomeMicrob Cell Graz Austria 6:356–369Google ScholarGong XWang SYu QWang MGe FLi SYu X2023Cla4 phosphorylates histone methyltransferase Set1 to prevent its degradation by the APC/CCdh1 complexSci Adv 9:eadi7238Google ScholarGrainger RJBeggs JD2005Prp8 protein: at the heart of the spliceosomeRNA N Y N 11:533–557Google ScholarGreen DMMarfatia KACrafton EBZhang XCheng XCorbett AH2002Nab2p is required for poly(A) RNA export in Saccharomyces cerevisiae and is regulated by arginine methylation via Hmt1pJ Biol Chem 277:7752–7760Google ScholarGuccione EBassi CCasadio FMartinato FCesaroni MSchuchlautz HLüscher BAmati B2007Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusiveNature 449:933–937Google ScholarGuillemette BDrogaris PLin H-HSArmstrong HHiragami-Hamada KImhof ABonneil ÉThibault PVerreault AFestenstein RJ2011H3 Lysine 4 Is Acetylated at Active Gene Promoters and Is Regulated by H3 Lysine 4 MethylationPLOS Genet 7:1–16Google ScholarGuy MPPodyma BMPreston MAShaheen HHKrivos KLLimbach PAHopper AKPhizicky EM2012Yeast Trm7 interacts with distinct proteins for critical modifications of the tRNAPhe anticodon loopRNA N Y N 18:1921–1933Google ScholarHalbach AZhang HWengi AJablonska ZGruber IMLHalbeisen REDehé P-MKemmeren PHolstege FGéli Vet al2009Cotranslational assembly of the yeast SET1C histone methyltransferase complexEMBO J 28:2959–2970Google ScholarHamey JJRakow SBouchard CSenst JMKolb PBauer UWilkins MRHart-Smith G2021Systematic investigation of PRMT6 substrate recognition reveals broad specificity with a preference for an RG motif or basic and bulky residuesFebs J 288:5668–5691Google ScholarHannich JTLewis AKroetz MBLi S-JHeide HEmili AHochstrasser M2005Defining the SUMO-modified proteome by multiple approaches in Saccharomyces cerevisiaeJ Biol Chem 280:4102–4110Google ScholarHe CLiu NXie DLiu YXiao YLi F2019Structural basis for histone H3K4me3 recognition by the N-terminal domain of the PHD finger protein Spp1Biochem J 476:1957–1973Google ScholarHenke RMDastidar RGShah ACadinu DYao XHooda JZhang L2011Hypoxia elicits broad and systematic changes in protein subcellular localizationAm J Physiol Cell Physiol 301:C913–928Google ScholarHirschhorn JNBrown SAClark CDWinston F1992Evidence that SNF2/SWI2 and SNF5 activate transcription in yeast by altering chromatin structureGenes Dev 6:2288–2298Google ScholarHowe FSBoubriak ISale MJNair AClynes DGrijzenhout AMurray SCWoloszczuk RMellor J2014Lysine acetylation controls local protein conformation by influencing proline isomerizationMol Cell 55:733–744Google ScholarHsu PLLi HLau H-TLeonen CDhall AOng S-EChatterjee CZheng N2018Crystal Structure of the COMPASS H3K4 Methyltransferase Catalytic ModuleCell 174:1106–1116Google ScholarHsu PLShi HLeonen CKang JChatterjee CZheng N2019Structural Basis of H2B Ubiquitination-Dependent H3K4 Methylation by COMPASSMol Cell 76:712–723Google ScholarHuang FChandrasekharan MBChen Y-CBhaskara SHiebert SWSun Z-W2010The JmjN domain of Jhd2 is important for its protein stability, and the plant homeodomain (PHD) finger mediates its chromatin association independent of H3K4 methylationJ Biol Chem 285:24548–24561Google ScholarHyllus DStein CSchnabel KSchiltz EImhof ADou YHsieh JBauer U-M2007PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylationGenes Dev 21:3369–3380Google ScholarIwase MToh-e A2001Nis1 encoded by YNL078W: a new neck protein of Saccharomyces cerevisiaeGenes Genet Syst 76:335–343Google ScholarJackson CAYadav NMin SLi JMilliman EJQu JChen YCYu MC2012Proteomic analysis of interactors for yeast protein arginine methyltransferase Hmt1 reveals novel substrate and insights into additional biological rolesProteomics 12:3304–3314Google ScholarJanke CMagiera MMRathfelder NTaxis CReber SMaekawa HMoreno-Borchart ADoenges GSchwob ESchiebel Eet al2004A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettesYeast 21:947–962Google ScholarJeon JMcGinty RKMuir TWKim J-AKim J2018Crosstalk among Set1 complex subunits involved in H2B ubiquitylation-dependent H3K4 methylationNucleic Acids Res 46:11129–11143Google ScholarJezek MSun WNegesse MYSmith ZMOrosz AGreen EM2023Set1 regulates telomere function via H3K4 methylation-dependent and -independent pathways and calibrates the abundance of telomere maintenance factorsMol Biol Cell 34:ar6Google ScholarKaczmarek Michaels KMohd Mostafa SRuiz Capella JMoore CL2020Regulation of alternative polyadenylation in the yeast Saccharomyces cerevisiae by histone H3K4 and H3K36 methyltransferasesNucleic Acids Res 48:5407–5425Google ScholarKan JZou LZhang JWu RWang ZLiang C2008Origin Recognition Complex (ORC) Mediates Histone 3 Lysine 4 Methylation through Cooperation with Spp1 in Saccharomyces cerevisiaeJ Biol Chem 283:33803–33807Google ScholarKarányi ZHalász LAcquaviva LJónás DHetey SBoros-Oláh BPeng FChen DKlein FGéli Vet al2018Nuclear dynamics of the Set1C subunit Spp1 prepares meiotic recombination sites for break formationJ Cell Biol 217:3398–3415Google ScholarKim JKim J-AMcGinty RKNguyen UTTMuir TWAllis CDRoeder RG2013The n-SET Domain of Set1 Regulates H2B Ubiquitylation-Dependent H3K4 MethylationMol Cell 49:1121–1133Google ScholarKim JRoeder RG2011Nucleosomal H2B ubiquitylation with purified factorsMethods San Diego Calif 54:331–338Google ScholarKim J-HSaraf AFlorens LWashburn MWorkman JL2010Gcn5 regulates the dissociation of SWI/SNF from chromatin by acetylation of Swi2/Snf2Genes Dev 24:2766–2771Google ScholarKim MVasiljeva LRando OJZhelkovsky AMoore CBuratowski S2006Distinct pathways for snoRNA and mRNA terminationMol Cell 24:723–734Google ScholarKim TBuratowski S2009Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5’ transcribed regionsCell 137:259–272Google ScholarKirmizis ASantos-Rosa HPenkett CJSinger MAGreen RDKouzarides T2009Distinct transcriptional outputs associated with mono- and dimethylated histone H3 arginine 2Nat Struct Mol Biol 16:449–451Google ScholarKirmizis ASantos-Rosa HPenkett CJSinger MAVermeulen MMann MBähler JGreen RDKouzarides T2007Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylationNature 449:928–932Google Scholarde La Roche Saint-André CGéli V2021Set1-dependent H3K4 methylation becomes critical for limiting DNA damage in response to changes in S-phase dynamics in Saccharomyces cerevisiaeDNA Repair 105:103159Google ScholarLaribee RNShibata YMersman DPCollins SRKemmeren PRoguev AWeissman JSBriggs SDKrogan NJStrahl BD2007CCR4/NOT complex associates with the proteasome and regulates histone methylationProc Natl Acad Sci U S A 104:5836–5841Google ScholarLee DCAitchison JD1999Kap104p-mediated nuclear import. Nuclear localization signals in mRNA-binding proteins and the role of Ran and RnaJ Biol Chem 274:29031–29037Google ScholarLee KWang K2018Small Noncoding RNAs in Agrobacterium tumefaciensCurr Top Microbiol Immunol 418:195–213Google ScholarLee KYChopra ABurke GLChen ZGreenblatt JFBiggar KKMeneghini MD2020A crucial RNA-binding lysine residue in the Nab3 RRM domain undergoes SET1 and SET3-responsive methylationNucleic Acids Res 48:2897–2911Google ScholarLee Y-SMulugu SYork JDO’Shea EK2007Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphatesScience 316:109–112Google ScholarLees NDSkaggs BKirsch DRBard M1995Cloning of the late genes in the ergosterol biosynthetic pathway of Saccharomyces cerevisiae--a reviewLipids 30:221–226Google ScholarLi H-TGong TZhou ZLiu Y-TCao XHe YChen CDZhou J-Q2015aYeast Hmt1 catalyses asymmetric dimethylation of histone H3 arginine 2 in vitroBiochem J 467:507–515Google ScholarLi NZhai YZhang YLi WYang MLei JTye B-KGao N2015bStructure of the eukaryotic MCM complex at 3.8 ÅNature 524:186–191Google ScholarLiang GKlose RJGardner KEZhang Y2007Yeast Jhd2p is a histone H3 Lys4 trimethyl demethylaseNat Struct Mol Biol 14:243–245Google ScholarLiu HYChiang YCPan JChen JSalvadore CAudino DCBadarinarayana VPalaniswamy VAnderson BDenis CL2001Characterization of CAF4 and CAF16 reveals a functional connection between the CCR4-NOT complex and a subset of SRB proteins of the RNA polymerase II holoenzymeJ Biol Chem 276:7541–7548Google ScholarLiu JLiu JStråby KB1998Point and deletion mutations eliminate one or both methyl group transfers catalysed by the yeast TRM1 encoded tRNA (m22G26)dimethyltransferaseNucleic Acids Res 26:5102–5108Google ScholarLow JKKWilkins MR2012Protein arginine methylation in Saccharomyces cerevisiaeFebs J 279:4423–4443Google ScholarLuciano PJeon JEl-Kaoutari AChallal DBonnet ABarucco MCandelli TJourquin FLesage PKim Jet al2017Binding to RNA regulates Set1 functionCell Discov 3:17040Google ScholarMargaritis TOreal VBrabers NMaestroni LVitaliano-Prunier ABenschop JJvan Hooff Svan Leenen DDargemont CGéli Vet al2012Two distinct repressive mechanisms for histone 3 lysine 4 methylation through promoting 3’-end antisense transcriptionPLoS Genet 8:e1002952Google ScholarMcBride AECook JTStemmler EARutledge KLMcGrath KARubens JA2005Arginine methylation of yeast mRNA-binding protein Npl3 directly affects its function, nuclear export, and intranuclear protein interactionsJ Biol Chem 280:30888–30898Google ScholarMei QXu CGogol MTang JChen WYu XWorkman JLLi S2019Set1-catalyzed H3K4 trimethylation antagonizes the HIR/Asf1/Rtt106 repressor complex to promote histone gene expression and chronological life spanNucleic Acids Res 47:3434–3449Google ScholarMeitinger FKhmelinskii AMorlot SKurtulmus BPalani SAndres-Pons AHub BKnop MCharvin GPereira G2014A memory system of negative polarity cues prevents replicative agingCell 159:1056–1069Google ScholarMendoza-Ochoa GIBarrass JDMaudlin IEBeggs JD2019Blocking late stages of splicing quickly limits pre-spliceosome assembly in vivoRNA Biol 16:1775–1784Google ScholarMersman DPDu H-NFingerman IMSouth PFBriggs SD2009Polyubiquitination of the demethylase Jhd2 controls histone methylation and gene expressionGenes Dev 23:951–962Google ScholarMiller TKrogan NJDover JErdjument-Bromage HTempst PJohnston MGreenblatt JFShilatifard A2001COMPASS: a complex of proteins associated with a trithorax-related SET domain proteinProc Natl Acad Sci U S A 98:12902–12907Google ScholarMizuno TNakamura MIrie K2018Induction of Ptp2 and Cmp2 protein phosphatases is crucial for the adaptive response to ER stress in Saccharomyces cerevisiaeSci Rep 8:13078Google ScholarMulder KWBrenkman ABInagaki Avan den Broek NJFTimmers HTM2007Regulation of histone H3K4 tri-methylation and PAF complex recruitment by the Ccr4-Not complexNucleic Acids Res 35:2428–2439Google ScholarMullen JRChen C-FBrill SJ2010Wss1 is a SUMO-dependent isopeptidase that interacts genetically with the Slx5-Slx8 SUMO-targeted ubiquitin ligaseMol Cell Biol 30:3737–3748Google ScholarMulugu SBai WFridy PCBastidas RJOtto JCDollins DEHaystead TARibeiro AAYork JD2007A conserved family of enzymes that phosphorylate inositol hexakisphosphateScience 316:106–109Google ScholarMurray SCHaenni SHowe FSFischl HChocian KNair AMellor J2015Sense and antisense transcription are associated with distinct chromatin architectures across genesNucleic Acids Res 43:7823–7837Google ScholarNadal-Ribelles MMas GMillán-Zambrano GSolé CAmmerer GChávez SPosas Fde Nadal E2015H3K4 monomethylation dictates nucleosome dynamics and chromatin remodeling at stress-responsive genesNucleic Acids Res 43:4937–4949Google ScholarNagy PLGriesenbeck JKornberg RDCleary ML2002A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3Proc Natl Acad Sci U S A 99:90–94Google ScholarNakagawa TKolodner RD2002The MER3 DNA helicase catalyzes the unwinding of holliday junctionsJ Biol Chem 277:28019–28024Google ScholarNayak AViale-Bouroncle SMorsczeck CMuller S2014The SUMO-specific isopeptidase SENP3 regulates MLL1/MLL2 methyltransferase complexes and controls osteogenic differentiationMol Cell 55:47–58Google ScholarNedea EHe XKim MPootoolal JZhong GCanadien VHughes TBuratowski SMoore CLGreenblatt J2003Organization and Function of APT, a Subcomplex of the Yeast Cleavage and Polyadenylation Factor Involved in the Formation of mRNA and Small Nucleolar RNA 3’-EndsJ Biol Chem 278:33000–33010Google ScholarNedea ENalbant DXia DTheoharis NTSuter BRichardson CJTatchell KKislinger TGreenblatt JFNagy PL2008The Glc7 Phosphatase Subunit of the Cleavage and Polyadenylation Factor Is Essential for Transcription Termination on snoRNA GenesMol Cell 29:577–587Google ScholarNg HHRobert FYoung RAStruhl K2003Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activityMol Cell 11:709–719Google ScholarNie MVashisht AAWohlschlegel JABoddy MN2015High Confidence Fission Yeast SUMO Conjugates Identified by Tandem Denaturing Affinity PurificationSci Rep 5:14389Google ScholarNiño CAGuet DGay ABrutus SJourquin FMendiratta SSalamero JGéli VDargemont C2016Posttranslational marks control architectural and functional plasticity of the nuclear pore complex basketJ Cell Biol 212:167–180Google ScholarNislow CRay EPillus L1997SET1, a yeast member of the trithorax family, functions in transcriptional silencing and diverse cellular processesMol Biol Cell 8:2421–2436Google ScholarNoma AKirino YIkeuchi YSuzuki T2006Biosynthesis of wybutosine, a hyper-modified nucleoside in eukaryotic phenylalanine tRNAEMBO J 25:2142–2154Google ScholarOki MNishimoto T1998A protein required for nuclear-protein import, Mog1p, directly interacts with GTP-Gsp1p, the Saccharomyces cerevisiae ran homologueProc Natl Acad Sci U S A 95:15388–15393Google ScholarOliete-Calvo PSerrano-Quílez JNuño-Cabanes CPérez-Martínez MESoares LMDichtl BBuratowski SPérez-Ortín JERodríguez-Navarro S2018A role for Mog1 in H2Bub1 and H3K4me3 regulation affecting RNAPII transcription and mRNA exportEMBO Rep 19:e45992Google ScholarPal SVishwanath SNErdjument-Bromage HTempst PSif S2004Human SWI/SNF-Associated PRMT5 Methylates Histone H3 Arginine 8 and Negatively Regulates Expression of ST7 and NM23 Tumor Suppressor GenesMol Cell Biol 24:9630–9645Google ScholarPemberton LFRosenblum JSBlobel G1997A distinct and parallel pathway for the nuclear import of an mRNA-binding proteinJ Cell Biol 139:1645–1653Google ScholarPerez AMThorner J2019Septin-associated proteins Aim44 and Nis1 traffic between the bud neck and the nucleus in the yeast Saccharomyces cerevisiaeCytoskelet Hoboken NJ 76:15–32Google ScholarPoornima GSrivastava GRoy BKuttanda IAKurbah IRajyaguru PI2021RGG-motif containing mRNA export factor Gbp2 acts as a translation repressorRNA Biol 18:2342–2353Google ScholarQu QTakahashi Y-HYang YHu HZhang YBrunzelle JSCouture J-FShilatifard ASkiniotis G2018Structure and Conformational Dynamics of a COMPASS Histone H3K4 Methyltransferase ComplexCell 174:1117–1126Google ScholarRadman-Livaja MLiu CLFriedman NSchreiber SLRando OJ2010Replication and Active Demethylation Represent Partially Overlapping Mechanisms for Erasure of H3K4me3 in Budding YeastPLoS Genet 6:e1000837Google ScholarRain JCSelig LDe Reuse HBattaglia VReverdy CSimon SLenzen GPetel FWojcik JSchächter Vet al2001The protein-protein interaction map of Helicobacter pyloriNature 409:211–215Google ScholarRizzardi LFDorn ESStrahl BDCook JG2012DNA replication origin function is promoted by H3K4 di-methylation in Saccharomyces cerevisiaeGenetics 192:371–384Google ScholarRoguev ASchaft DShevchenko APijnappel WWWilm MAasland RStewart AF2001The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4EMBO J 20:7137–7148Google ScholarRuthenburg AJAllis CDWysocka J2007Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic markMol Cell 25:15–30Google ScholarSantos-Rosa HBannister AJDehe PMGéli VKouzarides T2004Methylation of H3 lysine 4 at euchromatin promotes Sir3p association with heterochromatinJ Biol Chem 279:47506–47512Google ScholarSantos-Rosa HMillán-Zambrano GHan NLeonardi TKlimontova MNasiscionyte SPandolfini LTzelepis KBartke TKouzarides T2021Methylation of histone H3 at lysine 37 by Set1 and Set2 prevents spurious DNA replicationMol Cell 81:2793–2807Google ScholarSayou CMillán-Zambrano GSantos-Rosa HPetfalski ERobson SHouseley JKouzarides TTollervey D2017RNA Binding by Histone Methyltransferases Set1 and Set2Mol Cell Biol 37:e00165–17Google ScholarSchlichter ACairns BR2005Histone trimethylation by Set1 is coordinated by the RRM, autoinhibitory, and catalytic domainsEMBO J 24:1222–1231Google ScholarSchulze JMJackson JNakanishi SGardner JMHentrich THaug JJohnston MJaspersen SLKobor MSShilatifard A2009Linking cell cycle to histone modifications: SBF and H2B monoubiquitination machinery and cell-cycle regulation of H3K79 dimethylationMol Cell 35:626–641Google ScholarSenger BSimos GBischoff FRPodtelejnikov AMann MHurt E1998Mtr10p functions as a nuclear import receptor for the mRNA-binding protein Npl3pEMBO J 17:2196–2207Google ScholarSerra-Cardona ADuan SYu CZhang Z2022H3K4me3 recognition by the COMPASS complex facilitates the restoration of this histone mark following DNA replicationSci Adv 8:eabm6246Google ScholarShin JAChoi ESKim HSHo JCYWatts FZPark SDJang YK2005SUMO modification is involved in the maintenance of heterochromatin stability in fission yeastMol Cell 19:817–828Google ScholarSmith D-LErce MALai Y-WTomasetig FHart-Smith GHamey JJWilkins MR2020Crosstalk of Phosphorylation and Arginine Methylation in Disordered SRGG Repeats of Saccharomycescerevisiae Fibrillarin and Its Association with Nucleolar LocalizationJ Mol Biol 432:448–466Google ScholarSoares LMBuratowski S2012Yeast Swd2 Is Essential Because of Antagonism between Set1 Histone Methyltransferase Complex and APT (Associated with Pta1) Termination FactorJ Biol Chem 287:15219–15231Google ScholarSoares LMHe PCChun YSuh HKim TBuratowski S2017Determinants of Histone H3K4 Methylation PatternsMol Cell 68:773–785Google ScholarSollier JLin WSoustelle CSuhre KNicolas AGéli Vde La Roche Saint-André C2004Set1 is required for meiotic S-phase onset, double-strand break formation and middle gene expressionEMBO J 23:1957–1967Google ScholarSommermeyer VBéneut CChaplais ESerrentino MEBorde V2013Spp1, a member of the Set1 Complex, promotes meiotic DSB formation in promoters by tethering histone H3K4 methylation sites to chromosome axesMol Cell 49:43–54Google ScholarSoniat MSampathkumar PCollett GGizzi ASBanu RNBhosle RCChamala SChowdhury SFiser AGlenn ASet al2013Crystal structure of human Karyopherin β2 bound to the PY-NLS of Saccharomyces cerevisiae Nab2J Struct Funct Genomics 14:31–35Google ScholarSouth PFHarmeyer KMSerratore NDBriggs SD2013H3K4 methyltransferase Set1 is involved in maintenance of ergosterol homeostasis and resistance to Brefeldin AProc Natl Acad Sci U S A 110:E1016–25Google ScholarSüel KEGu HChook YM2008Modular organization and combinatorial energetics of proline-tyrosine nuclear localization signalsPLoS Biol 6:e137Google ScholarSun Z-WAllis CD2002Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeastNature 418:104–108Google ScholarTakeo KIto T2017Subcellular localization of VIP1 is regulated by phosphorylation and 14-3-3 proteinsFEBS Lett 591:1972–1981Google ScholarTerzi NChurchman LSVasiljeva LWeissman JBuratowski S2011H3K4 Trimethylation by Set1 Promotes Efficient Termination by the Nrd1-Nab3-Sen1 PathwayMol Cell Biol 31:3569–3583Google ScholarThandapani PO’Connor TRBailey TLRichard S2013Defining the RGG/RG motifMol Cell 50:613–623Google ScholarThornton JLWestfield GHTakahashi Y-HCook MGao XWoodfin ARLee J-SMorgan MAJackson JSmith ERet al2014Context dependency of Set1/COMPASS-mediated histone H3 Lys4 trimethylationGenes Dev 28:115–120Google ScholarTrahan COeffinger M2022Single-Step Affinity Purification (ssAP) and Mass Spectrometry of Macromolecular Complexes in the Yeast S. cerevisiaeMethods Mol Biol Clifton NJ 2477:195–223Google ScholarTravesa AKuo Dde Bruin RAMKalashnikova TIGuaderrama MThai KAslanian ASmolka MBYates JRIdeker Tet al2012DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1EMBO J 31:1811–1822Google ScholarTrésaugues LDehé P-MGuérois RRodriguez-Gil AVarlet ISalah PPamblanco Mpierre LUCIANOQuevillon-Cheruel SSollier Jet al2006Structural Characterization of Set1 RNA Recognition Motifs and their Role in Histone H3 Lysine 4 MethylationJ Mol Biol 359:1170–1181Google ScholarVitaliano-Prunier AMenant AHobeika MGéli VGwizdek CDargemont C2008Ubiquitylation of the COMPASS component Swd2 links H2B ubiquitylation to H3K4 trimethylationNat Cell Biol 10:1365–1371Google ScholarWalter DMatter AFahrenkrog B2014Loss of histone H3 methylation at lysine 4 triggers apoptosis in Saccharomyces cerevisiaePLoS Genet 10:e1004095Google ScholarWang LCollings CKZhao ZCozzolino KAMa QLiang KMarshall SASze CCHashizume RSavas JNet al2017A cytoplasmic COMPASS is necessary for cell survival and triple-negative breast cancer pathogenesis by regulating metabolismGenes Dev 31:2056–2066Google ScholarWang YDing ZLiu XBao YHuang MWong CCLHong XCong Y2018Architecture and subunit arrangement of the complete Saccharomyces cerevisiae COMPASS complexSci Rep 8:17405Google ScholarWarnecke DCHeinz E1994Purification of a Membrane-Bound UDP-Glucose:Sterol [beta]-D-Glucosyltransferase Based on Its Solubility in Diethyl EtherPlant Physiol 105:1067–1073Google ScholarWeiner AChen HVLiu CLRahat AKlien ASoares LGudipati MPfeffner JRegev ABuratowski Set al2012Systematic Dissection of Roles for Chromatin Regulators in a Yeast Stress ResponsePLoS Biol 10:e1001369Google ScholarWill CLLührmann R2011Spliceosome structure and functionCold Spring Harb Perspect Biol 3:a003707Google ScholarWilson TEPadgett KAJohnston MMilbrandt J1993A genetic method for defining DNA-binding domains: application to the nuclear receptor NGFI-BProc Natl Acad Sci U S A 90:9186–9190Google ScholarWorley JLuo XCapaldi AP2013Inositol pyrophosphates regulate cell growth and the environmental stress response by activating the HDAC Rpd3LCell Rep 3:1476–1482Google ScholarWykoff DDO’Shea EK2005Identification of sumoylated proteins by systematic immunoprecipitation of the budding yeast proteomeMol Cell Proteomics MCP 4:73–83Google ScholarXu DFarmer AChook YM2010Recognition of nuclear targeting signals by Karyopherin-β proteinsCurr Opin Struct Biol 20:782–790Google ScholarYoshida KBlobel G2001The karyopherin Kap142p/Msn5p mediates nuclear import and nuclear export of different cargo proteinsJ Cell Biol 152:729–740Google ScholarYoshimura SHHirano T2016HEAT repeats - versatile arrays of amphiphilic helices working in crowded environments?J Cell Sci 129:3963–3970Google ScholarYuan C-CMatthews AGWJin YChen CFChapman BAOhsumi TKGlass KCKutateladze TGBorowsky MLStruhl Ket al2012Histone H3R2 Symmetric Dimethylation and Histone H3K4 Trimethylation Are Tightly Correlated in Eukaryotic GenomesCell Rep 1:83–90Google ScholarZhang ZShibahara KStillman B2000PCNA connects DNA replication to epigenetic inheritance in yeastNature 408:221–225Google ScholarArticle and author informationAuthor informationPierre Luciano*Marseille Cancer Research Center (CRCM), U1068 Inserm, UMR7258 CNRS, Aix Marseille University, Institut Paoli-Calmettes, Marseille, France. Equipe labellisée LigueORCID iD: 0000-0002-1394-4129*Joint authorsKihyun Park*Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea*Joint authorsStéphane AudebertMarseille Cancer Research Center (CRCM), U1068 Inserm, UMR7258 CNRS, Aix Marseille University, Institut Paoli-Calmettes, Marseille, France. Equipe labellisée LigueORCID iD: 0000-0002-9409-2588Luc CamoinMarseille Cancer Research Center (CRCM), U1068 Inserm, UMR7258 CNRS, Aix Marseille University, Institut Paoli-Calmettes, Marseille, France. Equipe labellisée LigueORCID iD: 0000-0002-1230-4787Carlos A Niño†Marseille Cancer Research Center (CRCM), U1068 Inserm, UMR7258 CNRS, Aix Marseille University, Institut Paoli-Calmettes, Marseille, France. Equipe labellisée Ligue†Present address: Carlos A. Niño, IFOM ETS, The AIRC Institute of Molecular Oncology, Milan, Italy.Da Kyeong ParkOchang Institute of Biological and Environmental Science, Korea Basic Science Institute, Cheongju, Republic of KoreaIsabella E Maudlin‡Wellcome Centre for Cell Biology and Institute of Cell Biology, School of Biological Sciences, University of Edinburgh, Edinburgh, United Kingdom‡Present address: Isabella E. Maudlin, Sir William Dunn School of Pathology, University of Oxford, Oxford, OX1 3RE, UK.Marion Dubarry§Marseille Cancer Research Center (CRCM), U1068 Inserm, UMR7258 CNRS, Aix Marseille University, Institut Paoli-Calmettes, Marseille, France. Equipe labellisée Ligue§Present address: Marion Dubarry, Univ Lyon, Université Claude Bernard Lyon 1, INSA-Lyon, CNRS, UMR5240, Microbiologie, Adaptation et Pathogénie, 10 rue Raphaël Dubois, F-69622, Villeurbanne, FranceLara LeeMarseille Cancer Research Center (CRCM), U1068 Inserm, UMR7258 CNRS, Aix Marseille University, Institut Paoli-Calmettes, Marseille, France. Equipe labellisée LigueMarlene OeffingerInstitut de recherches cliniques de Montréal, Center for Genetic and Neurological Diseases, Montréal, Canada, Département de biochimie et médecine moléculaire, Faculté de Médicine, Université de Montréal, Québec, Canada, Division of Experimental Medicine, Faculty of Medicine, McGill University, Montréal, CanadaJean D BeggsWellcome Centre for Cell Biology and Institute of Cell Biology, School of Biological Sciences, University of Edinburgh, Edinburgh, United KingdomYoung Hye KimOchang Institute of Biological and Environmental Science, Korea Basic Science Institute, Cheongju, Republic of KoreaJaehoon KimDepartment of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon, Republic of KoreaFor correspondence: kimjaehoon@kaist.eduBernhard DichtlSchool of Life and Environmental Sciences, Deakin University, Geelong, AustraliaFor correspondence: bernhard.dichtl@deakin.edu.auVincent Géli¶Marseille Cancer Research Center (CRCM), U1068 Inserm, UMR7258 CNRS, Aix Marseille University, Institut Paoli-Calmettes, Marseille, France. Equipe labellisée LigueORCID iD: 0000-0002-4103-7462For correspondence: vincent.geli@inserm.fr¶Present address: Vincent Géli, Institute for Research on Cancer and Aging, Nice (IRCAN), Nice, FranceAuthor NotesCompeting interests: No competing interests declaredVersion historySent for peer review: November 23, 2025Preprint posted: November 29, 2025Reviewed Preprint version 1: March 6, 2026Cite all versionsYou can cite all versions using the DOI https://doi.org/10.7554/eLife.109886. This DOI represents all versions, and will always resolve to the latest one.Copyright© 2026, Luciano et al.This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.Metricsviews0downloads0citations0Views, downloads and citations are aggregated across all versions of this paper published by eLife.