Cell Stem Cell:北京大学赵扬团队系统解析“在体重编程”诱导心肌再生的关键病理障碍

专属客服号

微信订阅号
大数据治理
全面提升数据价值
赋能业务提质增效


编辑丨王多鱼
排版丨水成文
心肌梗死是全球主要死亡原因之一,且发病率呈逐年上升趋势,对患者生命造成重大威胁。由于成年哺乳动物心脏的再生能力极其有限,心梗区域的心肌因缺血坏死后取而代之的是心脏纤维化等病理过程。利用重编程因子将心梗区激活态心脏成纤维细胞“在体”原位诱导成为功能性心肌细胞,有望同时抑制心脏纤维化及实现心肌高效再生,是再生医学领域的梦想。但“在体重编程”较之培养皿中诱导重编程的难度更大。
北京大学赵扬研究组近年来深耕“在体重编程”的调控,报道了一系列可以提高心梗区激活成纤维细胞重编程效率的新因子和小分子,和周斌研究组合作利用严格的谱系示踪系统对其进行系统观测和验证(Circulation, 2023; iScience, 2023;Front Cell Dev Biol, 2020);以及进一步揭示心梗微环境中免疫细胞(巨噬细胞、T 细胞)及心脏纤维化相关因子等调控在体重编程的分子机制(Protein & Cell, 2024;Life Medicine, 2026)。然而,复杂的病理微环境因素仍像一道道无形的屏障使得在体重编程仍远达不到体外重编程的效果;而传统的研究方法也只能先逐一提出候选因子假设,再依次开展基于谱系示踪体系的动物实验验证;每个候选因子的在体研究都耗费大量时间和资源,并且难以同批对比大量不同候选因子。这也阻碍了“在体重编程”调控因子的快速解析及这一再生医疗策略从概念验证走向转化应用。
2026 年 3 月 30 日,北京大学赵扬研究组(北京大学未来技术学院分子医学研究所博士生蔡依泓、博士后杨洋、博士生杨峻博为论文共同第一作者)在 Cell Stem Cell 期刊发表了题为:Perturb-seq uncovers pathological obstacles to direct cardiac reprogramming in vivo 的研究论文。
研究团队建立了一套专为研究在体复杂病理环境而设计的在体细胞命运扰动图谱构建方案,绘制了心梗区域“在体重编程”诱导心肌再生的细胞命运转化图谱,并从 140 个候选因子中锁定了首要障碍因子——钙网蛋白(Calreticulin,CALR)。
该研究发现,心梗区域的应激响应激活 CALR 的表达,使其在内质网中大量驻留 Ca2+,降低胞质钙浓度和钙信号活性,进而抑制了关键重编程因子 MEF2C 的活性。通过抑制 CALR 或激活钙离子信号活性,能够显著提升心肌重编程效率和诱导心肌细胞的质量。Calr 敲低结合重编程因子,能够促进心梗后的在体重编程效率,促进心脏功能恢复,从而为心梗的再生医疗提供了理论基础和有转化前景的新靶点。

“精准导航”——在体重编程诱导心肌再生的细胞命运扰动图谱
为了精准定位在体重编程的层层“路障”,批量、系统地鉴定阻碍在体心脏重编程的关键因子,赵扬团队建立了一套研究在体重编程的 Perturb-seq 技术流程。他们首先把目标范围缩小在 131 个候选基因上——它们在病理环境中的表达量高于体外培养并被 WGCNA 分析判断为节点基因,并同时加上 9 个已报道因子作为参照。随后,将这 140 个因子的 shRNA(每个候选因子选用 2 条 shRNA,每条 shRNA 载体都带有唯一的分子标签)随着重编程因子和荧光蛋白阵列式地转导进入激活态心脏成纤维细胞内。研究团队进一步把这些“同时植入多个重编程因子和不同分子标签”的成纤维细胞混合移植进入另几只小鼠的心梗区域,用 Dox 诱导重编程因子的表达,并分别在诱导重编程1周和2周时间点分离表达荧光蛋白的细胞样品进行单细胞全转录组联合分子标签的 RNA 测序,进而成功构建了一张贯穿在体重编程全过程的“细胞命运扰动图谱”。“阵列式感染+细胞移植”的设计确保了在体重编程因子递送非常充分,且每种“扰动”的细胞比例也更为平均。
通过单细胞 RNA 测序分析,研究团队首先系统性地绘制了从成纤维细胞向心肌细胞命运及其它亚型转化的细胞命运图谱。研究团队发现,在心肌梗死的病理背景下,仅有小部分细胞向心肌命运演进。大量成纤维细胞由于微环境的干扰而陷入“病理”状态,如特定亚群高表达干扰素响应基因。通过对这一细胞命运图谱的深入解析,研究人员得以在分子水平上定义重编程过程中的不同细胞状态,为后续寻找拦截细胞命运转化的“幕后黑手”提供了高精度的细胞命运座标系。
通过分析扰动因子对应分子标签的分布,这一图谱清晰地展示了不同扰动因子对细胞命运走向的调控作用。通过计算每一个扰动因子在命运轨迹上的富集程度,研究人员发现,抑制特定的基因能够显著改变细胞的演化方向,使其绕过病理阻碍、更高效地诱导特定细胞亚群,包括诱导心肌细胞。这个图谱还提供了将这 100 多个因子进行头对头对比的机会,可以针对任意细胞群排序候选调控因子的作用效果。
“罪魁祸首”——CALR,成纤维细胞向心肌转分化的“刹车片”
在这一信息含量丰富的扰动图谱中,钙网蛋白(CALR,由 Calr 基因编码)在心肌样细胞群中高度富集,被鉴定为阻碍心肌样细胞亚群产生的首要障碍因子。CALR 通常定位于内质网,调控钙离子的储存和蛋白质折叠,同时也可以定位于胞质,是递呈给巨噬细胞的“吃我”信号。研究团队发现,在心肌梗死区域的成纤维细胞中,CALR 的表达水平显著升高,且这一现象在人类心梗患者的样本中同样存在。这意味着,CALR 的激活表达作为细胞应激后的反应,却可能在“无意中”成为了心肌再生的“刹车片”。
上述猜想通过体内、外功能实验得以验证。研究团队验证了敲低 Calr 能显著提升体内和体外场景下的重编程诱导效率。更令人振奋的是,这种提升不仅体现在数量上,更体现在质量上:Calr 的敲低使得诱导性心肌细胞在体外展现出前所未有的同步钙信号振荡,这意味着它们在功能上更接近功能性心肌细胞。此外,Calr 敲低联合其他重编程因子的共同作用下,原本极难被诱导为心肌的激活态心脏成纤维细胞实现了近 70% 的心肌诱导效率。在体重编程实验也显示,Calr 敲低也同样提高心肌样细胞的在体诱导效率达一个数量级,达到 10-20%。且在人类心脏成纤维细胞上,Calr 敲低也同样大幅提高重编程效率。
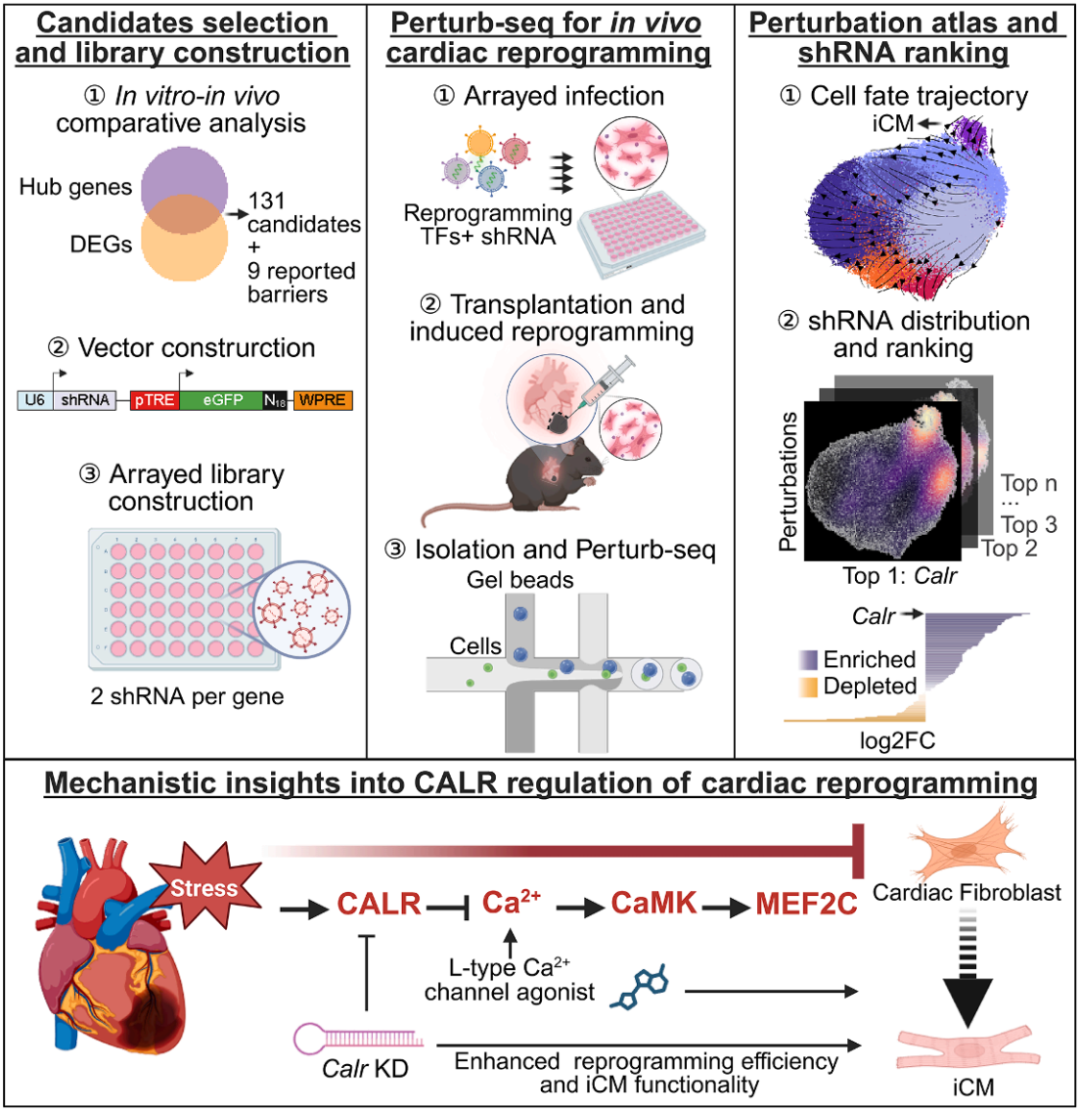
图1. 基于细胞移植的在体 Perturb-seq 揭示“在体重编程”首要障碍因子 CALR
“机制揭秘”——病理应激激活 Calr 通过 Ca2+信号调控 MEF2C 活性
那么,CALR 是如何调控成纤维细胞向心肌命运的重编程呢?
研究团队分析了多个线索,逐步将其定位于 Ca2+信号通路的调控上。敲低 CALR 会导致内质网向胞质释放 Ca2+,引起胞质内 Ca2+ 浓度的阶段性提升。这种钙信号的大幅提升激活了下游的 CaM/CaMKIIδ 通路。最为关键的是,无论是 Calr 敲低还是通过 L 型 Ca2+通道激动剂提升胞内 Ca2+浓度,都显著增强了重编程核心转录因子 MEF2C 的转录活性,解释了它们大幅提升重编程效率的原因。在某种程度上,Calr 敲低或者 Ca2+ 激活甚至可以替代外源 MEF2C,借助心脏成纤维细胞中原本就内源低表达的 MEF2C,并通过加强其活性来驱动成纤维细胞跨越命运鸿沟,顺利向心肌细胞转化。这一机制还将胚胎时期的心肌发育机制与病理环境下的再生过程联系了起来,证明了“重启”发育程序是实现高效再生的有效途径。
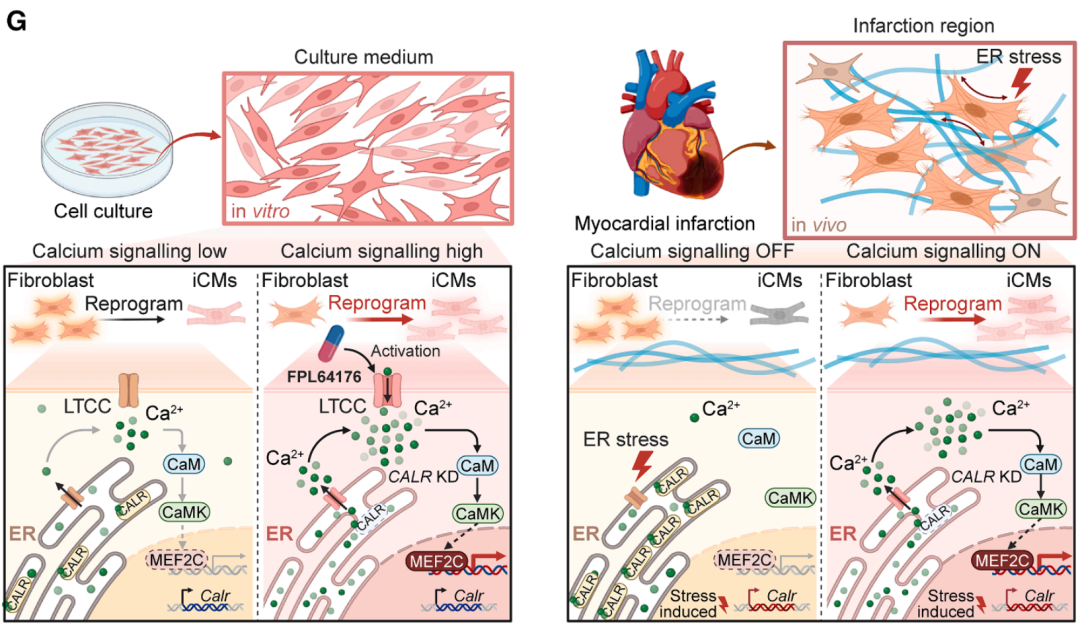
图2. 应激信号通过调控钙离子浓度的时空调控影响心脏重编程的机制解析
技术革新与未来展望
该研究是首次将 Perturb-seq 应用于“在体重编程”的研究,为“在体重编程”领域寻找更多调控靶点提供了新的技术框架,提示了直接在病理环境下研究在体重编程的重要性。尤其该研究绘制了心脏成纤维细胞向心肌以及多个成纤维细胞亚群之间命运转化的调控因子图谱;对大量候选因子进行了在体重编程效果的“头对头”对比;揭示了 CALR 在“在体重编程”诱导心脏修复再生中的核心作用及其通过调控 Ca2+ 通路进而作用于细胞命运核心转录因子活性的分子机制。这一进展为治疗心梗“在体重编程”策略提供了坚实的科学依据,并提供了具有临床应用潜力的新靶点。
论文链接:
https://doi.org/10.1016/j.stem.2026.03.006








点在看,传递你的品味

