Cell Death & Differ:中山大学蔡木炎/谢丹等团队揭示胃癌化疗耐药新机制

专属客服号

微信订阅号
大数据治理
全面提升数据价值
赋能业务提质增效
BTAF1是一种ATP依赖性的TBP-DNA复合体重塑因子,在胃癌中频繁发生突变。然而,其在DNA修复中的作用及治疗相关性仍不甚明确。
2026年3月17日,中山大学蔡木炎,谢丹和Jin-Ling Duan共同通讯在
Cell Death & Differentiation
在线发表题为
BTAF1: a key regulator of DNA end resection and predictor of chemotherapy sensitivity in gastric cancer
的研究论文。
该研究揭示,BTAF1基因敲除会通过损害同源重组(HR)修复的DNA末端切除过程,导致双链断裂(DSB)积累,从而在体外和体内均增强细胞对基因毒性药物的敏感性。从机制上讲,BTAF1能够阻止MRE11发生泛素化介导的降解,维持其蛋白稳定性,进而促进DNA末端切除与HR修复,最终增强细胞对DNA损伤应激的抵抗能力。
值得注意的是,在DNA损伤应答过程中,BTAF1与MRE11之间的相互作用受到PARP1介导的BTAF1 PAR酰化修饰的动态调控。BTAF1缺失也会增加胃癌异种移植模型和类器官模型的化学治疗敏感性。临床分析表明,在接受新辅助化学治疗的胃癌患者中,BTAF1高表达与不良预后相关。综上所述,该研究结果证实BTAF1通过稳定MRE11成为HR修复的关键调控因子,并提出BTAF1可作为预测基因毒性化学治疗反应的潜在生物标志物。

基因组不稳定性是癌症的一个标志性特征,其源于DNA损伤的累积,并使肿瘤细胞对基因毒性药物尤为敏感。为在内源性和外源性应激下维持基因组完整性,细胞依赖于一个高度协调的DNA损伤应答(DDR)网络。
在不同类型的DNA损伤中,双链断裂(DSB)具有最强的细胞毒性,主要通过易错的非同源末端连接(NHEJ)或高保真的同源重组(HR)进行修复。作为最精确的修复途径,HR对于维持基因组稳定性和抑制肿瘤发生不可或缺。
基因毒性化学治疗药物——包括铂类化合物、拓扑异构酶抑制剂和烷化剂——主要通过诱导DSB发挥其细胞毒性效应。HR缺陷(常由BRCA1/2突变导致)会增加肿瘤对这些药物的敏感性,并构成了基于PARP抑制剂的合成致死效应的机制基础。相反,HR的过度活化则驱动化学治疗耐药,其机制涉及HR效应因子(如RAD51)的上调或上游调控因子(包括ATR-CHK1轴)的失调。
这种适应性反应增强了DSB修复能力,导致对顺铂、依托泊苷及相关基因毒性药物的耐药,从而对实现持久疗效构成了重大挑战。因此,解析HR在介导化学治疗敏感性与耐药性中的双重作用,对于精准肿瘤学至关重要。
HR的一个关键早期步骤是DNA末端切除,该过程由MRE11-RAD50-NBS1(MRN)复合物启动,并由磷酸化的CtIP促进。作为MRN复合物的催化核心,MRE11具有核酸酶活性,可驱动短程末端切除并抑制NHEJ。MRE11的功能调控涉及多种翻译后修饰,包括泛素化、SUMO化、乳酸化和磷酸化。
值得注意的是,MRE11的失调已被证明会改变细胞对放射治疗和化学治疗的敏感性。然而,在DNA修复中调控MRE11蛋白稳定性及功能调节的机制在很大程度上仍未阐明。
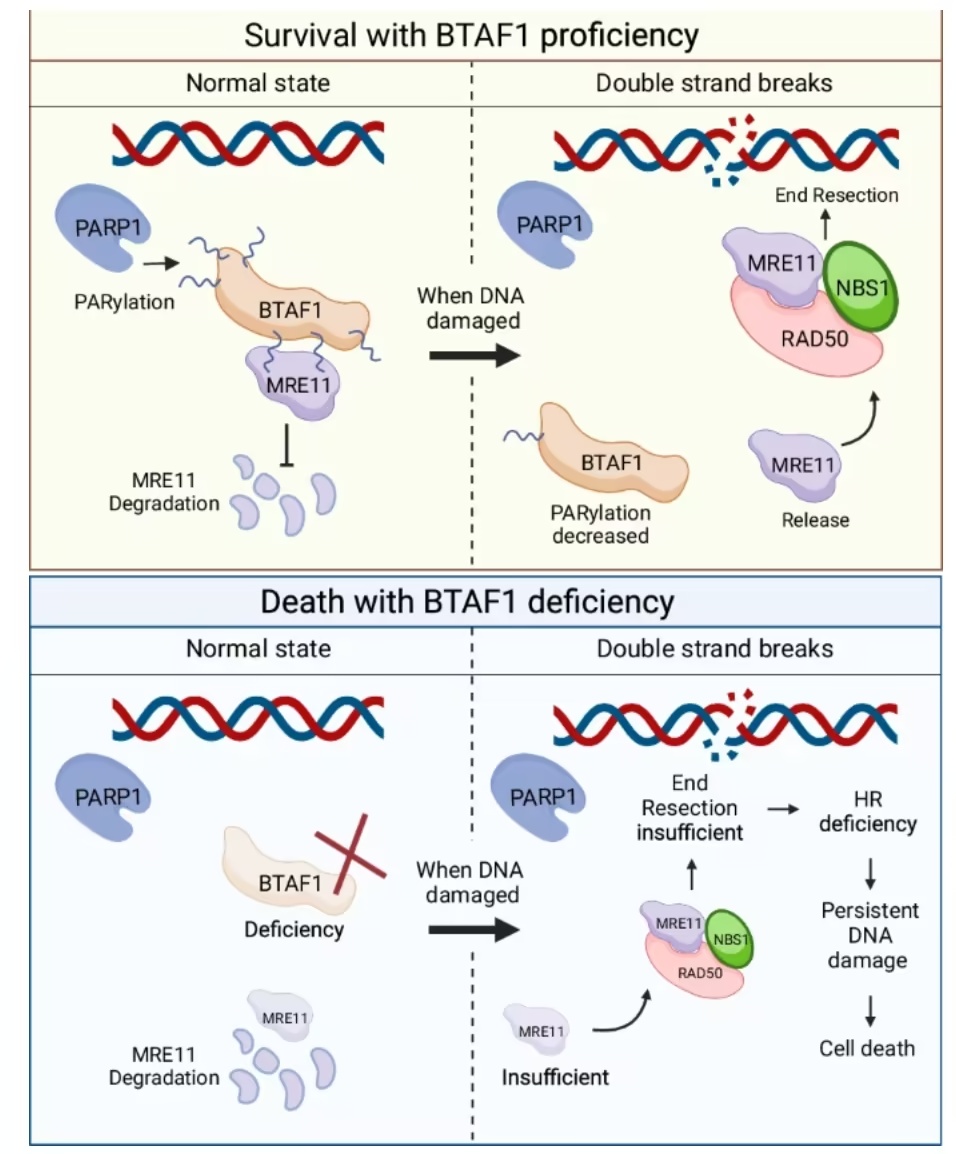
模式机理图(图片源自
Cell Death & Differentiation
)
BTAF1(B-TFIID TATA盒结合蛋白相关因子1)是一种保守的SWI2/SNF2家族ATP酶,它与TATA结合蛋白(TBP)形成稳定复合物,负向调控TBP与DNA的交互作用,从而影响RNA聚合酶II介导的转录起始。除转录调控外,BTAF1还参与染色质结构的调节。尽管具有这些重要功能,BTAF1在癌症发生发展和治疗应答中的生物学相关性仍很大程度上未被探索。
在本研究中,作者证明PARP1介导的BTAF1 PAR化增强了其与MRE11的交互作用,从而稳定MRE11并促进高效的DSB修复。在基因毒性应激下,BTAF1从PARP1上解离,以促进MRE11向染色质募集并加速DNA末端切除。BTAF1的缺失会增加对DNA损伤药物的敏感性。
临床分析表明,胃癌中BTAF1表达降低与更好的化学治疗应答和更有利的患者预后相关。总之,该研究结果强调了BTAF1作为MRE11“海绵”在调控DSB修复中的关键作用,并提出其可作为胃癌的潜在生物标志物和治疗靶点。
原文链接:https://www.nature.com/articles/s41418-026-01709-6

