Science:实验室闭环框架使复杂的多突变蛋白快速进化

专属客服号

微信订阅号
大数据治理
全面提升数据价值
赋能业务提质增效
蛋白质工程的搜索空间随着复杂性呈指数级增长。一个仅有100个氨基酸的蛋白质就有20
100
种可能的变体——比可观测宇宙中的原子数量还要多。传统的工程方法可能测试数百种变体,但将探索限制在序列空间的狭窄区域内。最近的机器学习方法通过计算筛选实现了更广泛的搜索。然而,这些方法仍然需要数万次测量,或者5-10轮迭代。

随着这些基础蛋白质模型的出现,蛋白质工程的瓶颈又回到了实验室。对于一个单一的蛋白质工程项目,研究人员只能有效地构建和测试数百种变体。如何选择这数百种变体,才能最有效地发现功能显著增强的进化蛋白质?为了解决这个问题,研究人员开发了MULTI-evolve,这是一个高效的蛋白质进化框架,它应用在约200种变体的数据集上训练的机器学习模型,这些变体特别专注于功能增强突变的成对组合。
这项发表在《科学》杂志上的工作,代表了Arc研究所首个用于生物设计的"实验室-循环"框架,其中计算预测和实验设计从一开始就紧密集成,反映了对人工智能引导研究的更广泛投入。
从成对相互作用中学习
进化蛋白质涉及两个基本步骤:找到有益的突变,然后将它们协同组合。在开发这种方法的早期,研究团队意识到,仅靠单突变数据训练的神经网络无法可靠预测哪些多突变组合会有效。这些模型缺乏关于突变如何相互作用的信息,而且大多数随机变体的大型数据集没有用处,因为绝大多数突变不会增强功能,因此测试数千个随机变体主要教会模型什么是无效的。
研究人员的见解是专注于质量而非数量;首先识别大约15-20个功能增强的突变(使用蛋白质语言模型或实验筛选),然后系统地测试所有这些有益突变的成对组合。这会产生100-200次测量,并且每一次测量都有助于学习有益的上位相互作用。
团队使用已发表研究中的12个现有蛋白质数据集对此进行了计算验证。仅用单突变和双突变数据训练神经网络,他们发现模型能够准确预测所有12个不同蛋白质家族中复杂的多突变体。即使将训练数据减少到可用数据的10%,这一结果依然成立。
在双突变体上训练之所以有效,是因为它们揭示了上位效应。一个双突变体可能表现优于其各部分之和、低于预期,或与预测完全一致。这些成对相互作用模式教会了模型突变组合的规则,使得模型能够外推预测哪些包含5、6或7个突变的组合将协同作用。
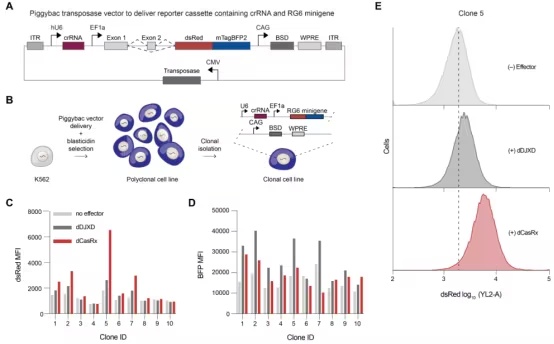
随后,团队将MULTI-evolve应用于三种新的蛋白质:APEX、用于反式剪接的dCasRx以及抗CD122抗体。对于dCasRx,他们从一个包含超过11,000种变体的深度突变扫描开始,仅提取功能增强的突变,并测试它们的成对组合——这证明了战略性数据筛选对于高效工程的价值。
每一项都只需在一轮实验中测试100-200种变体来训练模型,这些模型准确预测了复杂的多突变体,将传统上需要5-10轮迭代、耗时数月的过程压缩到了数周。
MULTI-evolve循环
MULTI-evolve将三项创新整合到一个端到端的框架中。
(1)组合蛋白质语言模型实现有效的突变发现。虽然单一突变可以改善蛋白质功能,但功能的显著提升需要组合多个突变。先前的工作已经证明蛋白质语言模型零样本方法能够预测哪些突变可能改善功能,但任何单一方法都只能识别出少数可用于生成高阶组合变体的突变。
为了识别许多功能增强的突变,团队的解决方案是组合来自几个不同模型的预测,一些分析蛋白质序列,另一些分析3D结构,并采用两种评分方法。在73个不同的蛋白质数据集上测试,他们发现他们的方法平均识别出约20个有益突变,而任何单一模型平均约11个。
当他们将这种方法应用于APEX时,识别出了A134P突变,该突变使活性提高了53倍。基于标准蛋白质语言模型的方法会系统地错过它,因为它们会惩罚脯氨酸替换。团队的一种集成评分策略涉及标准化氨基酸特异性偏差,比如这种针对脯氨酸替换的偏差,这使得A134P成为一个候选,否则它会被忽略。
(2)神经网络预测哪些组合效果最佳。下一步是确定,在获得一组有益的单突变体和成对双突变体后,将它们组合成包含多达七个突变的多突变变体的最有效方法。
通过计算基准测试,团队证明全连接神经网络能够通过主要在单突变和双突变体上训练来可靠地预测多突变体的活性。在12个不同的蛋白质数据集上,他们的模型有一半以上的时间正确识别了表现最佳的变体。
在实践中,他们证明MULTI-evolve能够识别出跨三种不同蛋白质、包含多达七个突变的超活性变体。他们通过一轮机器学习设计了多突变变体,模型在一个约200种战略性变体的紧凑训练集上训练,并且他们实验测试了少至九个提出的候选变体。
(3)MULTI-assembly方法实现快速合成。另一个瓶颈是构建和测试预测的变体。商业DNA合成昂贵且缓慢,特别是对于复杂的多突变体。现有的多点诱变实验室方法效率低,且寡核苷酸设计主观,可能导致结果不可靠。
为了解决这个问题,研究人员开发了MULTI-assembly,一种高效构建复杂变体的多点诱变方法。通过系统优化反应条件、寡核苷酸设计和组装参数,他们对于跨越数千碱基、包含多达九个突变的变体实现了40-70%的组装效率。他们还开发了一个计算寡核苷酸设计器,该设计器以目标突变为输入,并输出为高效组装优化的引物。所有这些都可以在数天而不是数周内完成。
亲自尝试MULTI-evolve
MULTI-evolve框架是模块化的,并将随着该领域的进步而改进。更好的蛋白质语言模型将增强突变发现,并且该方法自然地与其他设计工具集成,优化计算设计的蛋白质或治疗候选物。
团队已将MULTI-evolve作为开源工具提供,该工具处理蛋白质语言模型预测、神经网络训练和MULTI-assembly寡核苷酸设计。无论您是在研究酶、基因组编辑器还是治疗性蛋白质,该框架都提供了一条从初始突变到优化多突变体的系统路径。(生物谷Bioon.com)
参考文献:
Vincent Q. Tran et al.
Rapid directed evolution guided by protein language models and epistatic interactions
, Science (2026). DOI: 10.1126/science.aea1820.

